
共轭体系的分类及结构特征
2021-07-14 02:47朱秋华
大学化学 2021年6期
朱秋华
南方医科大学药学院,广州 510515
在有机化学中,共轭体系是指相邻原子或基团的电子轨道发生重叠或者说发生相互作用,电子发生离域运动、体系能量降低、键长平均化的结构单元。共轭体系的存在不仅对化合物的物理性质有很大影响,如对紫外吸收光谱、荧光光谱、核磁共振谱的影响,也对化合物的反应活性及反应发生位置有很大的影响。因此,共轭体系不仅是有机化学课程中一个非常重要的内容,也是理论化学、材料化学、分析化学、药物化学等多个领域研究的一个重要内容。共轭体系对化合物性质的影响源于其独特的电子云密度分布变化或者说电子效应–共轭效应。不同原子或基团组成的共轭体系具有不同类型和程度的共轭效应。共轭效应是有机化学课程的重点和难点内容,掌握共轭体系的分类及结构特征,对理解化合物的共轭效应具有重要作用。但一般的有机化学教材及文献对此没有系统的介绍。一般大学有机化学教材只是在讨论1,3-丁二烯独特的结构与性质时,简单地把共轭体系定义为:键长平均化,共轭体系中的各原子共平面并有一个垂直于该平面的p轨道,以及各原子上的p轨道互相平行并重叠的电子流域结构体系。而把碳氢σ单键(σCH)与π键或p轨道重叠而形成的电子流域体系称为超共轭体系。随着共轭体系的深入研究,共轭体系的内涵已远远超出了一般大学有机化学教材对共轭体系的定义,如文献报道的空间共轭[1,2]、含饱和碳原子的芳香体系[3]、碳-氟σ单键(σCF)参与的超共轭[4]等。本文综述了共轭体系的类型及结构特征,以拓展共轭体系在有机化学教学中的内涵,加深师生对共轭体系及共轭体系对化合物结构与性质影响的理解。因为共轭体系与π键和σ键有关,本文在介绍共轭体系的分类及结构特征前,先简单地介绍π键与σ键在价键(valence bond,VB)理论以及分子轨道(molecular orbital,MO)理论中的表示方法。然后通过实例的结构式、VB及MO理论的电子轨道示意图,阐明共轭体系的分类及其结构特征。
1 VB及MO理论中的共价键形成示意图
共轭体系一般是根据参与共轭的电子轨道类型来进行分类。为了更好地理解共轭体系的分类及结构特征,我们需要先了解一下VB及MO理论对共价键的定义及电子轨道示意图。VB理论认为共价键的本质是原子相互接近时,原子价层的电子轨道(n电子轨道)重叠(波函数叠加),原子间通过共用自旋相反的电子对,使能量降低而形成的化学键。原子轨道重叠具有方向性,轨道以头碰头方式重叠形成电子云呈键轴对称的σ键,以肩并肩方式重叠形成电子云呈面对称的π键。图1a为原子的n电子轨道(s、p和spx杂化电子轨道,x= 1–3)重叠形成σ键和π键的示意图。图中原子轨道重叠示意图和成键轨道图是σ键和π键的两种表示方法,常常在讨论反应机理、电子效应及共轭体系时使用。除了这两种表示方法外,还有大家熟悉的短横线“–”和电子对“˙˙”表示法。
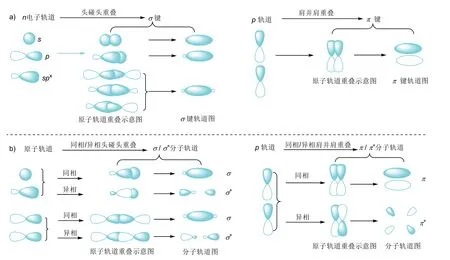
图1 VB理论的σ和π键(a)以及MO理论的成键/反键分子轨道(σ和π/σ*和π*)形成示意图(b)
MO理论认为原子形成分子后,电子不再属于个别的原子轨道,而是属于分子轨道。分子轨道是原子轨道的线性组合,有几个原子轨道就可以组合成几个分子轨道。类似VB理论中σ键和π键的形成,原子轨道以头碰头方式重叠形成电子云呈键轴对称的σ分子轨道,而原子轨道以肩并肩方式重叠形成电子云呈面对称的π分子轨道。形成分子轨道的原子轨道能量相近。波函数(原子轨道)同相重叠形成的分子轨道又称为成键分子轨道,用σ和π表示,而原子轨道异相重叠形成的分子轨道称为反键分子轨道,用σ*和π*表示。成键分子轨道能量比原子轨道能量低,反键分子轨道能量比原子轨道能量高。当电子进入成键分子轨道的数目比进入反键分子轨道的数目多时,能量降低,形成稳定的化学键。图1b为一个s轨道和一个杂化轨道同相/异相头碰头重叠形成σ/σ*分子轨道,两个p轨道同相/异相头碰头重叠形成σ/σ*分子轨道,以及两个p轨道同相与异相肩并肩重叠形成π/π*分子轨道的示意图。对比MO理论中的σ和π成键分子轨道与VB理论中的σ和π键,可以看到二者的轨道图及符号其实是相同的,只是名称和含义有所不同。
2 共轭体系的分类
报道的共轭体系分类有三种,即基于电子轨道类型、共轭体系中相邻原子或基团之间的连接方式以及电子轨道共轭程度大小的分类。
2.1 基于电子轨道类型的分类
相邻原子或基团的电子轨道发生重叠是共轭体系最根本的结构特征。因此,共轭体系的分类一般是根据电子轨道的类型进行分类。从VB理论来看,共轭体系是由相邻原子或基团的成键电子轨道(π键或σ键电子轨道)与成键或n电子轨道重叠所形成的能量降低结构体系;而从MO理论来看,共轭体系是由含一对电子的成键分子轨道(π或σ成键分子轨道)或n轨道与空的反键分子轨道(π*或σ*)或含0–1个电子的p轨道相互作用,形成能量更低的成键分子轨道和能量更高的反键分子轨道,进入能量更低的成键分子轨道电子数比进入能量更高的反键轨道电子数多,从而使能量降低的结构体系(共轭体系MO能级示意图如图2所示)。如表1所示,根据VB理论中的电子轨道类型,共轭体系可分为π–π、n–π、σ–π、σ–n和σ–σ五种类型,而根据MO理论中的电子轨道类型,可将共轭体系可分为π–π*、n–π*、σ–π*、π–p、n–p、σ–p、π–σ*、n–σ*和σ–σ*九种类型。
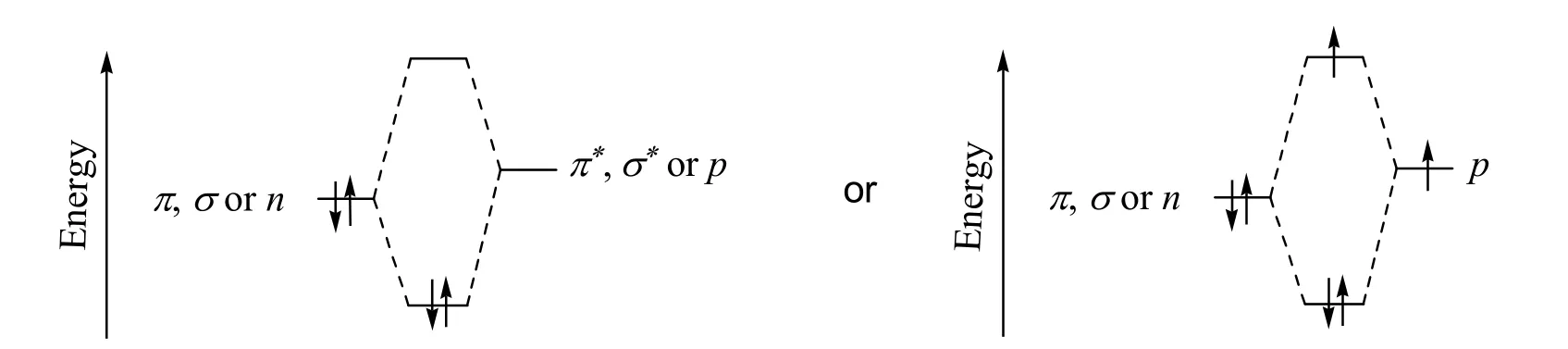
图2 形成共轭体系的MO能级示意图

表1 基于VB和MO理论中电子轨道类型的共轭体系分类
广义的共轭体系是指由各种电子轨道重叠所形成的电子离域的结构体系,但一般把无σ键轨道参与的共轭体系称为共轭体系,而把有σ键轨道参与的共轭体系称为超共轭体系,如Alabugin等[5]在超共轭综述中所述。根据VB理论中的轨道类型,共轭体系可分为π–π和n–π共轭,超共轭体系可分为σ–π、σ–n和σ–σ超共轭。而根据MO理论中的轨道类型,共轭体系分为π–π*、n–π*、π–p和n–p共轭,超共轭体系分为σ–π*、σ–p、σ–σ*、π–σ*和n–σ*超共轭。
2.2 基于相邻共轭基团连接方式的分类
共轭体系中相邻的原子或原子团可以是连在单键或双键两端的原子或基团、同一碳原子上的原子或基团、或者非价键直接相连但在空间上相邻的原子或基团。一般把非价键相连但在空间上相邻的原子或基团形成的共轭体系称为空间共轭(through-space conjugation),而把由价键相连的邻位或同位原子或基团形成的共轭体系称为价键共轭(through-bond conjugation)。
2.3 基于共轭效应强弱的分类
Mulliken等[6,7]认为饱和与不饱和基团参与的共轭,只有量的区别而没有性质的差别。建议将π与π键轨道的作用称为一级共轭,σ和π键的相互作用称之为二级共轭或者一级超共轭,而把σ键之间的作用称之为三级共轭或二级超共轭。
3 共轭体系的结构特征
由于在有机化学教材中只提到了基于电子轨道类型的共轭体系分类,而未涉及基于共轭体系中相邻原子或基团之间的连接方式以及电子轨道共轭程度大小的分类。并且,电子轨道的重叠是形成共轭体系的本质特性。因此,在以实例阐明不同共轭体系的结构特征时,以基于电子轨道类型分类的共轭体系,狭义共轭体系与超共轭体系,进行讨论。
3.1 狭义共轭体系的结构特征
狭义共轭体系,即无σ键参与的共轭体系,包含通过价键形成的π–π和n–π共轭体系,以及通过空间(through-space,TS)形成的π–π和n–π共轭体系(TS-π–π和TS-n–π共轭体系)。由于通过价键形成的共轭体系比较普遍,而通过空间形成的共轭体系只在特殊结构的分子中存在,在讨论体系类型时,除非指明是空间共轭,一般是指价键共轭。下面通过实例、简单明了的VB理论轨道示意图(见图1a,π键用原子轨道重叠示意图表示)阐明共轭体系的结构特征。
3.1.1π–π共轭体系结构特征
π–π共轭体系就是如图3所示的重键(双键或叁键)与单键间隔相连的分子结构单元。图3a是比较大的π–π共轭体系,图3b是最简单的π–π共轭体系。在这些最简单的共轭体系中,单键(标示红色的键)在中间像一根扁担一样两端各挑一个重键。1,3-丁二烯就是这样一个最简单的π–π共轭体系。如图3c和3d所示,当我们把1,3-丁二烯分子中的两个π键分别用两个肩并肩的p轨道表示时,就会发现,四个p轨道全部相邻、平行。如图3c所示,这四个p轨道是相互重叠的,其中1、2号碳上的两个p轨道以及3、4号碳上的两个p轨道的重叠程度大于2、3号碳上的两个p轨道的重叠程度。但我们经常用如图3d所示的,用相互平行但不重叠的p轨道来表示肩并肩重叠形成了π键的两个p轨道。实际上,只要相邻碳原子上有平行的p轨道,它们就会重叠并形成如电子云呈面对称的π键(见图1中π键电子云图)。也就是说,1,3-丁二烯分子形成了一个4原子4电子的大Π键(Π44)(Πnm,n为参与共轭的电子轨道数,m为参与共轭的电子数[8])。四个碳原子上的p轨道里各含一个电子,这4个p轨道里的4个电子在这个大Π键中做流域运动,而不是局限在1、2位和3、4位碳原子之间,即1、2位和3、4位碳原子之间的电子云密度降低,使1、2位和3、4位之间的C=C双键键长变长(0.137 nm,普通C=C双键键长0.133 nm),而2和3位碳原子之间电子云密度增加,使2和3位之间的C-C单键键长变短(0.146 nm,普通C-C单键键长0.154 nm)。也就是说,是这种大Π键的存在使1,3-丁二烯的键长平均化。我们可以想到,这个π–π共轭体系有多长,就可以形成多长的大Π键,即共轭体系里的大Π键电子就可以在多长的范围做离域运动。如图3a所示的苯环分子是一个环状的Π66共轭体系,其键长完全平均化(键长0.140 nm)。
另外,图3e从MO理论中的成键分子轨道与反键分子轨道的相互作用以及MO能级示意图解释了1,3-丁二烯分子中的π–π共轭作用。如图3e所示,分子中一个C=C之间的π成键分子轨道可以作为电子供体,而另一个C=C之间的π*反键分子轨道作为电子受体,π和π*分子轨道相互作用形成能量更低、运动范围更大的成键分子轨道,电子进入该分子轨道,从而使体系能量降低,键长平均化。值得一提的是,这种MO轨道对于π–π共轭的解释是把π–π共轭看成两个π键的相互作用[5]。如果把分子中四个原子的p轨道重叠当成一个大的Π键用MO理论来讨论,分子轨道示意图、MO能级图以及对π–π共轭体系能量降低,键长平均化的解释有所不同[9]。

图3 存在π-π共轭体系的几个分子结构简式(a和b)、以肩并肩平行的p轨道表示1,3-丁二烯(CH2=CH-CH=CH2)中π键的结构式(c和d)以及以π和π*分子轨道和MO能级示意图表示的π–π*共轭作用(e)
3.1.2n-π共轭体系结构特征
n–π共轭体系是相邻原子或基团的非键n电子轨道与π键电子轨道相互作用而形成的共轭体系。因为n电子轨道包含p轨道和spx杂化轨道,所以n–π共轭体系又可分为p–π和spx–π共轭。
3.1.2.1p–π共轭体系结构特征
p–π共轭体系是相邻原子或基团的p轨道与π键轨道相互作用形成的共轭体系。按照参与共轭的p轨道里含电子的多少,又可以把p–π共轭分成:含2个电子、1个电子和0个电子的p轨道与π键形成的p–π共轭体系。由于一个原子轨道只能容纳2个电子,含2个电子的p轨道可以称为满电子的轨道,而含1个电子和0个电子的p轨道可以分别称为单电子和无电子的空轨道,所以用p(满)–π、p(单)–π或者p(空)–π来分别表示含2个电子、1个电子和0个电子的p轨道与π键形成的p–π共轭体系。
如图4所示的溴乙烯分子/烯丙基负离子、烯丙基自由基、丙烯基正离子分别存在p(满)–π、p(单)–π或者p(空)–π共轭。Br原子价层有七个电子,其中一个电子与碳形成σ键后,还有三对未参与成键的电子,这些未成键的电子对也叫孤对电子,其中有两对孤对电子位于p轨道中。碳负离子、碳自由基及碳正离子是反应过程中C-X键发生异裂或均裂形成的活泼中间体,异裂或均裂后的碳原子杂化轨道转变成p轨道[9]。如图4b所示,当我们把这些化合物中的π键用两个肩并肩平行的p轨道来表示时,就可以很清楚地看到,溴原子/碳负离子、碳自由基及碳正离子上分别含一对电子、一个单电子和无电子的p轨道与π键中的p轨道相邻并平行,即重叠形成了电子离域的3原子4电子(Π34)、3原子3电子(Π33)和3原子2个电子(Π32)的大Π键共轭体系。

图4 存在p-π共轭体系的溴乙烯(BrCH=CH2)、烯丙基负离子(CH2=CHCH2-)、丙烯基自由基(CH2=CHCH2·)以及丙烯基正离子(CH2=CHCH2+)的结构式
3.1.2.2spx-π共轭体系结构特征
大家都知道,苯胺分子中N原子的碱性远小于脂肪胺中N原子的碱性,即苯胺分子中N原子上孤对电子接收质子的能力,远小于脂肪胺中N原子上孤对电子接收质子的能力。其原因是苯胺分子中N原子上的孤对电子与苯环中的π键形成了共轭。有机化学教材指出苯胺分子中N原子上的孤对电子是以介于sp2与sp3杂化轨道之间的杂化轨道参与共轭。可以肯定的是,无论是以什么样的杂化轨道参与共轭,N原子上参与共轭的非键孤对电子轨道不可能与共轭的π键p轨道平行(如图5a所示),即与有机化学教材中关于共轭体系中p轨道相互平行的定义是不相符的。显而易见,杂化轨道与相邻π键电子轨道的重叠程度要小于π–π、p–π共轭体系相邻p轨道的重叠程度。
我们的实验结果显示,与苯相连的N原子可以以介于sp2与sp3杂化轨道之间的杂化轨道成键,也可以以sp2杂化轨道成键。如图5b所示,在1,3-二苯基-4,5-二羧乙基四氢嘧啶(diethyl 1,2,3,6-tetrahydro-1,3-diphenylpyrimidine-4,5-dicarboxylate,TTHP)分子中,存在两个与苯基相连的氮原子,即N1和N2。N1与三个碳原子形成的σ键键角均小于sp2杂化轨道键角120°,但大于NH3中N原子以sp3杂化轨道成键的键角106.8°,这说明N1原子如有机化学教材中提到的,是以介于sp2与sp3之间的杂化轨道成键。但该分子中的N2与三个碳原子形成的σ键键角之和刚好等于360°,且N2与相连的三个碳原子共平面,这说明N2原子是以sp2杂化轨道成键。值得一提的是,该N2原子sp2杂化轨道平面与苯环平面成一定的夹角,而与连有酯基的C=C双键平面几乎共平面。这说明N2的孤对电子主要与连有酯基的C=C双键共轭。TTHP为我们发展的一个多组分反应产物,单晶结构发表在该合成方法文章中[10]。

图5 苯胺(C6H5NH2)分子中的spx–π共轭(a)及N原子以sp2 (N2)和介于sp2与sp3之间的杂化轨道(N1)成键的实例(b)
3.1.3 空间共轭体系结构特征
空间共轭体系(TS-共轭)是指共轭的原子或基团之间没有σ单键相连,但它们由于空间距离比较近,也像图3–5中以σ单键相连的原子或基团一样可以通过π与π或π与n轨道的重叠,形成电子流域的共轭体系。空间共轭不仅存在于分子内,也存在于分子间,即空间共轭可以分为分子内和分子间的TS-共轭体系。
3.1.3.1 分子内的TS-共轭体系结构特征
如图6中的化合物1[11]和化合物2[2],其空间相邻的苯基(红色虚线相连的苯基)π键电子轨道重叠,形成了π–π空间共轭(TS-π–π)。而在化合物 THP-1 (diethyl 1,2,3,6-tetrahydro-1,2,3-triphenylpyrimidine-4,5-dicarboxylate)中,其空间相邻的N原子上的孤对电子轨道与C=C中的π键电子轨道重叠,形成了n-π空间共轭体系(TS-n-π) (红色虚线相连的N原子和C=C双键)。

图6 几个分子内具有空间共轭的化合物
化合物的空间共轭可以从前线轨道电子云分布图反映出来。化合物THP-1是我们在2011年发展的一个高效的合成一系列新型 C6位未取代四氢嘧啶类化合物(C6-unsubstituted tetrahydropyrimidines, THPs)的多组分反应产物[12,13]。如图7所示,THP-1可以形成两种分别发蓝色荧光和绿色荧光的同质多晶(1b和1g)。虽然THP-1是共轭程度很低的非平面分子,但其荧光晶体THP-1b和THP-1g荧光量子率却分别高到72%和93%[14]。发光机理研究发现THP-1的分子中存在空间共轭[14]。如图7中最高占有分子轨道(HOMO)电子云分布图HOMO-1b (II)和-(III)所示,N1上含孤对电子的杂化轨道与C=C双键中的π键电子轨道在空间上相邻并重叠(红色圆圈标注的部分),形成了n–π空间共轭。N1原子上的三个共价键夹角分别为110.3°、117.5°和118.2° (conformation-1b-I),键角均小于120°,但大于106.8°,这说明以N1原子以介于sp2与sp3杂化轨道成键。

图7 THP-1的分子结构以及两种荧光异构体图(1b和1g) (365 nm荧光灯下)、1b单晶中不同角度的构象(conformation-1b I–III)及1b单晶构象的HOMO电子云分布图(HOMO-1b II–III)
3.1.3.2 分子间的TS-共轭体系结构特征
空间共轭不仅存在于分子内,还存在于分子间。香港科技大学唐本忠院士团队仔细研究了几个仅含游离苯基的小分子固态荧光产生机理,如研究了1,1,2,2-四苯基乙烷分子在溶液中的荧光发射光谱波长仅为290 nm,而固态的荧光发射光谱波长为476 nm的发光机理。实验和理论计算结果证明分子间苯基的π–π空间共轭是该化合物固态发光的主要光物理作用机制[15]。
吸收波长红移是产生共轭体系的一个典型特征。分子的平面化也能使吸收波长红移,但平面化引起的红移一般为20–30 nm,并且具有结构振动峰,而分子聚集引起的红移则往往比较大,并且精细结构峰消失[16]。THPs具有很强的聚集诱导发光(aggregation-induced emission,AIE)特性。AIE特性是2001年香港科技大学唐本忠院士课题组发现并命名的一种独特荧光特性,即,在溶液中不发光,但聚集时发光的荧光特性[17]。AIE克服了传统的荧光化合物在稀溶液中具有很高的荧光效率,但在浓溶液中或聚集时荧光减弱甚至淬灭的缺点,在多种应用领域中发挥了其独特的优势。目前已发现THPs可以用于活细胞内质网成像[18]、可擦写压致荧光变色材料[19]以及灵敏的测定表面活性剂临界胶束浓度的荧光点亮探针[20,21]等。THP-1b和1g还具有独特的在不同温度范围的荧光温度响应特性[22]。THPs的荧光特性不仅与其分子内的空间共轭有关,还与其分子间的电子相互作用有关。THP-1b和1g的激发波长比其在环己烷溶液中的吸收波长分别长38和92 nm,说明在THP-1b和1g中存在空间共轭。其在晶体结构中分子间芳基间的距离小于0.5 nm也证明了芳基π电子的相互作用[14],即存在空间共轭。
3.2 超共轭体系的结构特征
超共轭体系为σ键参与的共轭体系。自Mulliken[6]在1939年发现并提出碳氢σ键(σCH)参与的超共轭效应以来,超共轭体系得到了很大的发展[5,23,24]。除了传统教材中提到的σCH外,其他多种原子与碳形成的σ键(σCX)也能参与共轭。超共轭体系的结构远比共轭体系复杂。Alabugin和Manoharan[25]通过自然轨道(natural bond orbital,NBO)分析法计算了不同σCX键的供电子能力:(Al, Ga) >> Ge > As ≥Si > P > B > Se > H > C > S > Br > N > Cl > O > F (≥表示二者差别小于2.1 kJ˙mol−1,>>表示二者差别小于大于12.6 kJ·mol−1),并指出σCH与σCC键的供电子能力相差很微小,传统上高估了二者的差别,该顺序会随分子结构不同而有所变化。Alabugin和Zeidan[26]通过NBO分析法计算的σCX键接受电子的能力顺序为:Br (6.3) > Cl (6.2) > SH(1) (5.4) > F (5.1) > OH(1) (4.7) ≈ SH(2) (4.7) ≈ SeH (4.7) ≈ PH2(1)(4.6) ≈ AsH2(4.5) ≈ NH2(1) (4.5) > OH(2) (4.2) > PH2(2) (4.0) > NH2(2) (3.8) ≈ GeH3(3.8) > SiH3(3.6) >CH3(3.4) > H (3.2),这里的≈表示二者差别小于0.4 kJ·mol−1。
根据VB电子轨道类型,超共轭分为σ–π、σ–n和σ–σ三种(参见表1)。由于文献在说明超共轭体系的结构特征及性质时常常以MO轨道示意图加以说明。在下面阐明超共轭体系的实例中,除了用简单明了的VB理论轨道示意图(见图1a,σ键和π键用原子轨道重叠示意图表示)外,还同时给出MO轨道示意图(见图1b,成键分子轨道σ和π,以及反键分子轨道π*,用原子轨道重叠示意图表示;而反键分子轨道σ*用分子轨道图表示)。从MO理论来看,σCX键参与共轭时,即可作为电子供体,以σCX成键分子轨道参与共轭;又可作为电子受体,以σ*CX反键分子轨道参与共轭。那么σCX参与共轭时到底是作为电子供体,还是作为电子受体,取决于X原子的电负性以及轨道的电子云密度大小。在其他条件相同的情况下,与电负性较大的原子形成的σCX键作为电子受体的能力较大,而作为供体的能力较小;反之,则作为电子供体的能力较大,而作为受体的能力较小。
3.2.1σ-π超共轭体系结构特征
如图8所示,σCX键以单键(标示红色的单键)与双键相连时,可以形成σ–π超共轭体系。传统的有机化学教材只提到σCH–π超共轭,认为σCH能够产生共轭效应的原因是因为氢原子小不能屏蔽σ键电子云,从而使σ键电子轨道可以与相邻的π轨道重叠,形成超共轭。如图8a所示,丙烯分子中甲基上的3个σCH键与C=C双键之间有单键(标示红色的单键)相连,可以形成σCH–π超共轭。当我们把甲基上的一个σCH键以及π键用电子轨道表示时,就可以很清楚地看到σCH键与π键的p道相邻,即可以发生重叠(见图8a中VB理论电子轨道示意图)。但这种重叠是从侧面发生的,其重叠程度远比π–π、p–π共轭体系中电子轨道的重叠程度小。所以,超共轭体系中的电子离域程度比π–π、p–π共轭体系离域程度弱。从MO理论来看,此时的σCH键在共轭体系中是作为电子供体,以成键分子轨道σCH参与共轭,而π键则作为电子受体,以π*CC反键分子轨道参与共轭,形成σCH-π*CC超共轭(见图8a中MO理论电子轨道示意图)。σCH成键分子轨道与π*CC反键分子轨道相互作用形成能量更低的成键分子轨道和能量更高的反键分子轨道,两个电子进入能量更低的成键分子轨道,体系能量降低(见图8a中MO能级示意图)。
除σCH键可以形成超共轭外,已报道其他原子与碳形成的σ键也可以形成超共轭。如图8b所示,全氟代环丁烯醇和全氟代环戊烯醇分子中有四个碳氟σCF键与π键之间有单键(标示红色的单键)相连,可以形成σCF–π超共轭。一般来说,由于环张力的存在,四元环的稳定性比五元环的稳定性差。但全氟代环丁烯醇的稳定性却比全氟代环戊烯醇的稳定性大[4]。这是因为四元环中的σCF键与π键的空间距离比五元环中的σCF键与π键的空间距离更近(见图8b中VB理论电子轨道示意图),具有更大的重叠程度,即更大的共轭程度所致[4]。由于F原子电负性比较大,从MO理论来看,σCF键在共轭体系中作为电子受体,以σ*CF反键分子轨道参与共轭,而π键则作为电子供体,以πCC成键分子轨道参与共轭,形成πCC–σ*CF超共轭(见图8b中MO理论电子轨道示意图)。σ*CF反键分子轨道与πCC成键分子轨道相互作用形成能量更低和更高的两个分子轨道,两个电子进入能量更低的分子轨道,体系能量降低(见图8b中MO能级示意图)。

图8 分子中存在σ-π超共轭体系的化合物
3.2.2σ-n超共轭体系结构特征
σ–n超共轭体系为σCX键与非键n电子轨道(非键p轨道和杂化轨道)重叠形成的共轭体系。如图9a–d所示,乙基正离子(CH3CH2+)、乙基自由基(CH3CH2·)、2-氟代乙基负离子(CH2FCH2-)和氟代甲氨分子(FCH2NH2)分子中存在σ–n超共轭体系。从VB理论来看,这些分子中的σ–n超共轭体系的形成,是以单键(标示红色的单键)相连的邻位σCX键与n电子轨道相互重叠所致(见图9a–d的第2行)。从MO理论来看,在乙基正离子和乙基自由基中,缺电子的碳正离子和碳自由基价层上分别含0个及1个电子的p轨道作为电子受体参与共轭,而含满电子的成键分子轨道σCH作为电子供体参与共轭,形成σCH–p(空)或与σCH–p(单)超共轭(见图9a和b的第3行)。p轨道与σCH成键分子轨道相互作用形成能量更低和更高的两个分子轨道,电子先填入能量更低的分子轨道,然后再填入能量更高的分子轨道,填入更低能量分子轨道的电子数多于填入更高能量分子轨道的电子数,体系能量降低(见图9a和b的第4行)。但在2-氟代乙基负离子和氟代甲氨分子中,是含电负性大的F原子σCF键以反键分子轨道σ*CF作为电子受体参与共轭,而满电子的p轨道或sp3杂化轨道作为电子供体参与共轭[5],形成σ*CF–p或与σ*CF–sp3超共轭(见图9c和d的第3行)。σ*CF与p轨道或sp3杂化轨道相互作用形成能量更低和更高的两个分子轨道,一对电子填入能量更低的分子轨道,体系能量降低(见图9c和d的第4行)。

图9 分子中含σ–n超共轭体系的化合物
在有机化学中,一般认为取代环己烷的稳定构象是较大基团以e键相连的、空间位阻较小的椅式构象。但当有电负性较大的元素存在时,情况并非如此。如图10a中的2-氯四氢-2H-吡喃的稳定构象是氯原子位于a键位的椅式构象(I),而不是氯原子位于e键位的椅式构象(II),I式转变成II式的自由能为9.08 kJ·mol−1[27]。这是因为I式中氧原子上含孤对电子的p轨道与反键分子轨道σ*CCl几乎平行,重叠程度比较大,可以形成更低能量的分子轨道,即形成更稳定的p–σ*CCl超共轭体系。这种现象被称为异头效应(anomeric effect),是Edward[28]于1955年首次在吡喃糖中发现的。σ–n超共轭对反应活性也有很大影响。如图10b所示,由于σCSi比σCH具有更强的给电子能力,可以与碳正离子的空p轨道形成更稳定的σ–n超共轭体系,使得仅仅将H换成SiCH3的消除反应速率增大2.4 ×1012倍[29]。

图10 σ–n超共轭对分子结构及反应活性的影响
3.2.3σ-σ超共轭体系结构特征
σ–σ超共轭体系是指由于相邻的σ键电子轨道的重叠而形成的超共轭体系。如图11a所示,乙烷分子具有两个典型的构象,即能量最高的重叠式和能量最低的交叉式。有关交叉式构象比重叠式构象稳定的原因,有机化学教材上的解释是重叠式中非键氢原子之间的距离(229 pm)小于两个氢原子范德华半径之和(240 pm)[9](图11b)或者认为是重叠式中C―H键上的σ电子对的排斥力较大[30](图11c)。1939年,Mulliken[6]首次指出超共轭效应是乙烷交叉式更稳定的主要原因。2001年,Vojislava Pophristic和Lionel Goodman[31]指出交叉式构象中含电子的σCH成键分子轨道与空的σ*CH反成分子键轨道重叠形成了更稳定的分子轨道,即形成了σCH–σ*CH共轭体系,从而使交叉式构象更稳定(σCH–σ*CH轨道重叠图如图11d所示)。超共轭效应是乙烷交叉式更稳定的主要原因得到了一些实验及理论的支持[32]。

图11 乙烷分子(CH3CH3)的交叉式(staggered)和重叠式(eclipsed)构象
前面已经提到,从MO理论来看,σ键参与共轭时,与碳形成σ键的原子电负性越大,该σ键作为电子受体的能力越大,而所为电子供体的能力越弱;反之,则作为电子供体的能力越大,而所为电子受体的能力越弱。这就是说,当两个电负性相差比较大的原子与碳形成的σ键相邻时,电负性比较大的原子与碳形成的反键分子轨道σ*可以作为有利的电子受体(acceptor),用σ*CA表示,而电负性比较小的原子与碳形成的成键分子轨道σ可以作为有利的电子供体(donor),用σCD表示。σ*CA与σCD相互作用,会产生更强的σ–σ超共轭效应。如图12a和b中1,2-二氟乙烷[33]和3-氟-N,N-二甲基哌啶正离子[34]的I式构象比II式构象能量更低更稳定,以及图12c中Z-1,2-二氟乙烯比E-1,2-二氟乙烯能量更低更稳定[35]。这是因为,在能量更低的构象式或顺反异构式中,σ*CA与σCD可以发生重叠(见图12中第2行MO理论电子轨道示意图,D原子用绿色表示,A原子用蓝色表示),形成稳定的超共轭体系。而在能量较高的构象式或顺反异构式中,σ*CA与σCD的重叠很少,不能形成稳定的超共轭体系。
3.2.4 不同位置的超共轭体系结构特征
前面讨论的超共轭体系分类是基于轨道类型的分类。同样,超共轭还可以根据相邻的原子或基团之间是否有价键相连,把超共轭分为通过价键的超共轭和通过空间的超共轭。在无σ键参与的共轭体系中,价键共轭体系中的原子或原子团是由σ单键相连的邻位原子或原子团。但在超共轭体系中,价键超共轭体系中的原子或原子团之间除了是由σ单键相连的邻位原子或原子团外,还可以是以双键相连的邻位原子或基团(见图12c)。另外,连接同一碳原子上的两个原子或基团也可以形成超共轭。如图13a所示,化合物3中连接同一碳上的两个σCC单键可以形成超共轭,并且该化合物中存在的超共轭效应远大于乙烷分子交叉构象中两个σCH单键的超共轭效应(NBO法计算的σwing–σ*inner与σwing–σ*wing之间的轨道作用能分别为69.5 kJ·mol−1和83.3 kJ·mol−1,而乙烷分子中的σCH–σ*CH仅为12.1 kJ·mol−1)[36]。从13a中MO电子轨道示意图可以看到,同碳原子上的键角α越小,σ-σ*的轨道重叠程度越大,即超共轭效应越大。这就是为什么化合物3中σwing–σ*inner与σwing–σ*wing两种类型的超共轭效应比乙烷分子中σCH–σ*CH超共轭效应大的原因。

图12 σ–σ共轭对分子结构的影响
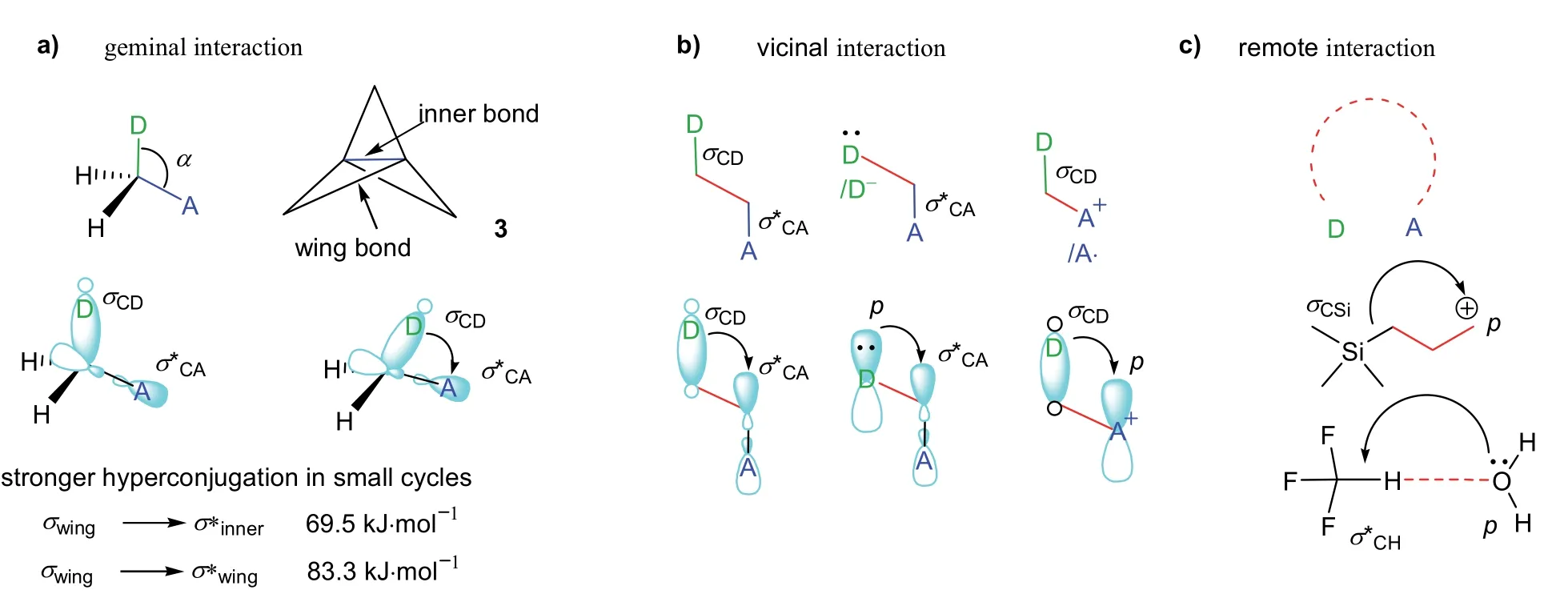
图13 基于超共轭体系中原子或原子团之间连接方式的超共轭体系分类[5]
空间超共轭体系中的原子或原子团的相对位置也比前面的空间共轭体系复杂。根据形成超共轭体系的原子或原子团是连接同一碳原子上的原子或原子团、还是连接在单键或双键两端的邻位原子或原子团、还是非直接相连的原子或原子团,Alabugin等[5]将超共轭体系分为同位(geminal)、邻位(vicinal)和远程(remote)原子或原子团轨道相互作用的超共轭体系(如图13a–c所示)。这里的远程超共轭类似前面讨论的空间共轭,可以发生在分子内,也可以发生在分子之间。分子内的远程超共轭体系可以是通过中间间隔多个价键的原子或基团。如图13c所示,在3-(三甲基硅烷基)丙基正离子中相隔两个σ键(标示红色的两个σ键)的碳正离子与C―Si基团发生σCSi―p超共轭[37]。远程超共轭体系也可以是分子间相邻的原子或基团。如图13c所示的三氟甲烷中的C―H基团与水分子中的氧原子之间形成的σCH―p超共轭[38]。
4 结语
综上所述,共轭体系是相邻原子或基团的电子轨道发生重叠,电子发生离域运动,键长平均化,体系能量降低的体系,可以存在于分子内和分子间,对化合物的结构、物理与化学性质有很大的影响。从上面的实例可以看出,相邻原子或基团的电子轨道重叠是共轭体系的本质特征。共轭体系可以根据电子轨道类型、相邻基团是否有价键相连以及共轭效应作用强度进行分类。广义的共轭体系包含所有电子轨道类型的共轭体系。但一般把无σ键参与的共轭体系称为共轭体系,而把有σ键参与的共轭体系称为超共轭体系。共轭和超共轭体系又可以根据参与共轭的轨道类型进行进一步分类(基于VB理论及MO理论中电子轨道的共轭体系分类见表1)。共轭和超共轭体系还可以根据共轭体系中相邻的原子或基团是否有价键相连,把共轭和超共轭体系分为价键共轭与空间共轭。空间共轭和超共轭又可分为分子内与分子间的空间共轭。一般文献或教材只是用VB或者MO理论的电子轨道图阐明共轭体系的分类及结构特征。本文通过实例,同时用VB和MO理论的电子轨道示意图阐明复杂的超共轭体系分类及结构特征,有助于从不同角度加深对共轭体系结构特征及共轭效应本质的理解。
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
原子与分子物理学报(2020年5期)2020-03-17
陕西中医(2018年6期)2018-08-29
考试周刊(2018年39期)2018-04-19
中国塑料(2016年2期)2016-06-15
中国塑料(2016年1期)2016-05-17
兽医导刊(2016年12期)2016-05-17
读写算·教研版(2016年8期)2016-05-07
中国塑料(2016年11期)2016-04-16
