
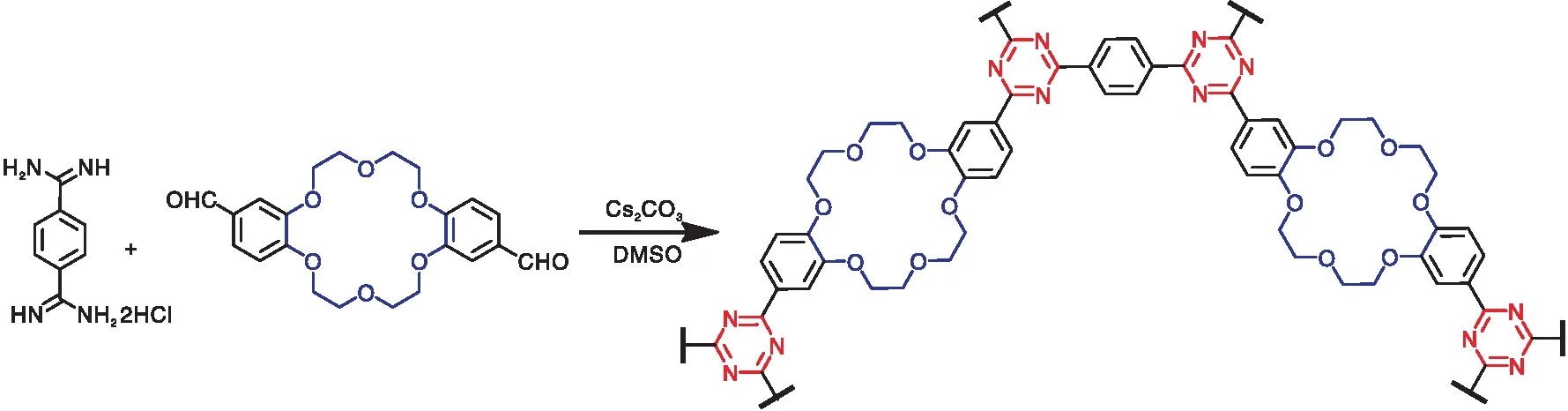
冠醚基共价三嗪框架促进二氧化碳催化转化为环状碳酸酯
2023-11-21 04:08陈可炽罗荣昌周贤太
工业催化 2023年10期
陈可炽,罗荣昌*,周贤太
(1.广东工业大学轻工化工学院,广东 广州 510006;2.中山大学惠州研究院,广东 惠州 516081)
化石燃料燃烧产生的过量CO2排放是全球面临的主要挑战之一[1]。与引起大众忧虑的常见空气污染物不同,CO2实际上是生产附加值产品不可或缺的C1原料,可以转化为碳酸酯、羧酸或甲醇等化学品[2]。CO2与环氧化物的环加成反应由于具有100%的原子经济效率,被认为是固定CO2最有吸引力的方法之一[3],截至目前,已经开发了一系列均相或多相催化剂,如碱金属盐、季鎓盐、离子液体、金属有机框架(MOFs)和多孔有机聚合物(POPs)等,应用于CO2与环氧化物环加成反应[4]。
POPs由于具有结构可调性、合成多样性以及易功能化等特点[5],被广泛研究并应用于吸附和催化领域。POPs大致可分为共轭微孔聚合物(CMPs)、自聚微孔聚合物(PIMs)、超交联聚合物(HCPs)、多孔芳香框架(PAFs)、共价三嗪框架(CTFs)和共价有机框架(COFs)等[6-7]。其中,CTFs由于其特殊的三嗪结构单元,具有富氮、高度稳定和多孔等特性,在CO2与环氧化物的环加成反应中具有广阔前途。CTFs的典型合成方法包括离子热法、超强酸催化、P2O5催化、醛脒缩合法和微波辅助法等。如Li Y M等[8]以2,5-二氰基吡啶为原料,采用ZnCl2催化的离子热三聚法在高温下合成了一种无金属CTFs,其催化活性归因于富氮的三嗪骨架和吡啶基团组成的碱性催化位。与其他方法相比,醛脒缩合法避免了反应温度过高、强酸或强碱的使用等系列问题,实现了在温和条件下一锅缩聚合成CTFs[9-10]。
碱金属盐作为一种廉价的环加成催化剂受到广泛关注。但其单独存在于反应体系时通常表现出较低的溶解度,限制了其催化活性[11-14]。通常将其与助催化剂搭配使用以克服溶解度低的缺点[15]。研究发现,冠醚由于具有独特的供电子空腔,与碱金属离子络合可以形成稳定的配合物,大幅度提高碱金属盐在反应体系中的溶解度[16-20]。Liu X等[21]发现,以反式二氨基二苯并-18-冠-6为功能结,用各种三醛型单体为连接剂,采用典型席夫碱缩合反应合成了系列冠醚功能化POPs材料。当原位螯合碘化钾后,在CO2与环氧化物的环加成反应中取得了较好的催化效果。我们猜想,将能够活化CO2的三嗪基团引入到冠醚型功能材料中有望进一步提高催化性能。然而,冠醚型CTFs还未有相关报道。
以二苯并-18-冠醚-6为基本模块,通过醛脒缩合反应在温和条件下制备得到一种冠醚型CTFs(命名为CE-CTF)。考虑尺寸效应的影响,CE-CTF中所含的冠醚结构对钾离子表现出优良的结合能力,使卤素阴离子的亲核性大幅度提高[22]。而原位形成的三嗪环结构又可以显著增强聚合物对CO2分子的吸附能力进而提高其催化性能[23-24]。因此,当使用KI作为助催化剂时,在相对温和的无溶剂条件下,CE-CTF催化材料在CO2催化转化为环状碳酸酯的反应中展现出优异的催化性能,归因于冠醚结构与三嗪基团协同催化作用。本文对冠醚基共价三嗪框架促进二氧化碳催化转化为环状碳酸酯进行研究。
1.实验部分
1.1 试 剂
二苯并-18-冠醚-6(98%)、四氢呋喃(99%)、乙腈(99.5%)、二氯甲烷(99%)、三氟乙酸(99%)、双(三甲基硅基)胺基锂溶液(1.0 mol·L-1)、乙酸乙酯(99.5%)、碘化钾(98%)、对苯二甲腈(98%),上海麦克林生化科技有限公司;六亚甲基四胺、盐酸、丙酮、二甲亚砜,分析纯,广州化学试剂厂;无水硫酸镁、无水乙醇、碳酸铯,分析纯,天津市致远化学试剂有限公司。
1.2 催化剂制备
1.2.1 反式-二醛基二苯并-18-冠-6的合成
反式-二醛基二苯并-18-冠-6的合成反应式为:

在惰性气体保护下,将二苯并-18-冠醚-6(1.006 g,2.42 mmol)和六亚甲基四胺(1.586 g,11.31 mmol)加入圆底烧瓶中,在0 ℃下缓慢滴加8 mL三氟乙酸,搅拌反应10 min后升温至120 ℃反应20 h,冷却至100 ℃后加入盐酸(3 mol·L-1,5 mL),搅拌30 min后冷至室温。最后用二氯甲烷萃取,用水洗涤后用无水硫酸镁干燥,过滤,真空脱除溶剂得到固体产物(产率70%)。1H NMR(400 MHz,DMSO-d6)δ 9.83(s,1H),7.54 (dd,J=8.2,1.8 Hz,1H),7.37(d,J=1.8 Hz,1H),7.16(d,J=8.3 Hz,1H),4.22-4.12(m,4H),3.89-3.84(m,4H)。
1.2.2 1,4-苯二羧酰胺盐酸盐的合成
1,4-苯二羧酰胺盐酸盐的合成反应式为:

将对苯二甲腈(4 g,25.6 mmol)溶于120 mL四氢呋喃中,0 ℃下于60 min内滴加双(三甲基硅基)胺基锂溶液(1 mol·L-1,120 mL),在25 ℃下混合物搅拌16 h,然后冷却至0 ℃。接着加入盐酸-乙醇(2 mol·L-1)进行猝灭,pH=8~9,把混合物放置过夜,过滤,用乙酸乙酯和四氢呋喃洗涤,在从水-乙醇混合物中重结晶得白色固体(产率65%)。1H NMR (400 MHz,DMSO-d6)δ 8.09(s,1H),7.93(s,2H),7.49(s,1H)。
1.2.3 CE-CTF的合成
将反式-二醛基二苯并-18-冠-6(416 mg,1.0 mmol)、1,4-苯二羧酰胺盐酸盐(470.4 mg,2.0 mmol)和碳酸铯(1.434 g,4.4 mmol)于圆底烧瓶中,加入二甲基亚砜(10 mL)和去离子水(0.4 mL),120 ℃反应12 h。反应后冷却至室温并用稀盐酸(3×10 mL)、水(3×10 mL)、丙酮(3×10 mL)和四氢呋喃(3×10 mL)洗涤过滤,得到的固体在70 ℃下真空干燥过夜,最后得到聚合物CE-CTF。
CE-CTF的合成反应式为:
1.3 催化剂表征
1H NMR(液体核磁)采用布鲁克400 MHz超导核磁共振仪AVANCE III HD 400测试制备的化合物。
采用Nicolet 6700型红外光谱仪进行傅里叶变换红外光谱(FT-IR)测试,(4 00~ 4 000) cm-1。
13C NMR(固体核磁)采用全数字超导核磁共振谱仪(NMR)600 MHz测试催化剂的化学结构。
采用场发射透射电子显微镜HT7700进行形貌测试。
采用比表面积仪和微孔物理吸附仪ASAP 2020对样品进行比表面积和孔径测试。
采用X射线光电子能谱Escalab 250Xi测试聚合物的表面元素组成。
热失重分析TGA采用STA449F5热重分析仪进行测试,样品在空气氛围下以10 ℃·min-1程序升温,测试温度范围:室温~800 ℃。
1.4 催化剂活性评价
将环氧氯丙烷(0.46 g,5.0 mmol)、CE-CTF(15 mg)和KI(5 mg)直接加入预干燥的10 mL不锈钢高压釜中。采用CO2对高压釜加压以保持所需压力,反应器迅速加热到所需温度。搅拌适当时间,在冰水浴中快速冷却高压釜,剩余的CO2气体缓慢释放。用乙酸乙酯提取目标产物,用气相色谱法测定产物的收率和选择性。催化剂通过过滤回收为固体,用甲醇和二氯甲烷等有机溶剂清洗多次,真空干燥,无需进一步纯化即可用于下一次运行。
2 结果与讨论
2.1 FT-IR
CE-CTF的FT-IR谱图如图1所示。
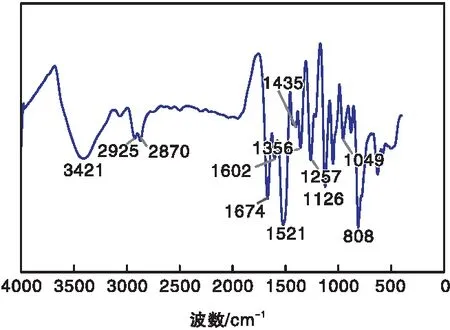
图1 CE-CTF的FT-IR谱图Figure 1 FT-IR spectrum of CE-CTF

2.2 固体核磁(Solid-state 13C MAR NMR)
CE-CTF的固体核磁碳谱图如图2所示。由图2可以看出,化学位移在70处的强峰是冠醚单元的亚甲基碳上的出峰,而化学位移在112~148处的一系列共振信号来源于苯环上的碳,其中化学位移在170处的峰为三嗪单元上碳的出峰,进一步证明CE-CTF的成功制备。

图2 CE-CTF的固体核磁碳谱图Figure 2 Solid-state 13C MAR NMR spectrum of CE-CTF
2.3 TEM
CE-CTF的TEM照片如图3所示。由图3可以看出,CE-CTF呈现出蜂窝状的堆叠状态,表明聚合物中纳米孔丰富,有助于CO2的吸附和物质传递。

图3 CE-CTF的TEM照片Figure 3 TEM image of CE-CTF
2.4 XPS
CE-CTF的XPS谱图如图4所示。由图4可以看出,CE-CTF中含有元素C、N和O。其中CE-CTF的N1s峰可以拟合成2个信号峰,结合能分别为398.4 eV和399.1 eV,对应于C=N和C-N基团,进一步证明聚合物中三嗪环的存在。而在CE-CTF的O1s峰中,对应于C-O-C的结合能峰出现在532.5 eV,表明冠醚基团存在于CE-CTF的骨架中。
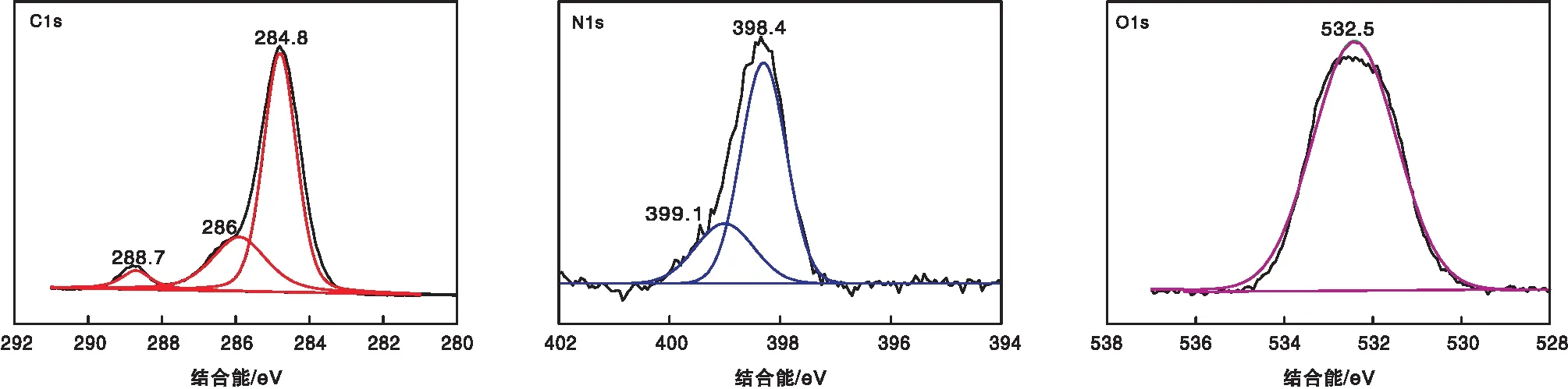
图4 CE-CTF的XPS谱图Figure 4 XPS spectra of CE-CTF
2.5 N2吸附-脱附
在-196 ℃和(0~0.1) MPa条件下进行N2吸附-脱附测试,结果如图5所示。由图5可以看出,相对压力<0.1时,氮气吸附量上升,表明该材料中存在一定可观数量的微孔;相对压力>0.1时,氮气吸附量平缓上升;相对压力>0.7时,氮气吸附量急剧上升,可判断该曲线为Ⅰ型曲线。CE-CTF的比表面积为10 m2·g-1,聚合物中离子的强极性产生了密集的堆叠结构导致其比表面积较低。尽管如此,聚合物主干中富氮三嗪结构的存在增强对CO2的捕获能力以提高其催化性能。

图5 CE-CTF的氮气吸附-脱附等温线Figure 5 N2 adsorption-desorption isotherm for CE-CTF
2.6 TGA
CE-CTF的TGA谱图如图6所示。由图6可以看出,样品在300 ℃前没有明显的分解,具有良好的热稳定性,有利于在CO2转化中的应用。

图6 CE-CTF的TGA谱图Figure 6 FT-IR spectrum of CE-CTF
2.7 催化性能测试
以CO2和环氧氯丙烷进行环加成反应为模型,在无添加任何溶剂和相对温和条件下,对不同催化剂在不同条件下的催化性能进行测试,在环氧氯丙烷(5 mmol)、催化剂+助催化剂(15 mg+5 mg)、CO2压力(1.0 MPa)和反应温度(80~120) ℃条件下,环氧氯丙烷与CO2在单体及催化剂上的环加成反应结果如表1所示。环加成反应式为:

表1 环氧氯丙烷与CO2在单体及催化剂上的环加成反应结果Table 1 Results of cycloaddition reaction ofepichlorohydrin with CO2 on monomer and catalyst

由表1可以看出,在100 ℃和CO2压力1.0 MPa下,不使用催化剂时反应无法发生。单独使用KI作为催化剂时,环状碳酸酯产率基本可以忽略,这可能是由于KI在环氧化物中的溶解性很低所致。当直接使用CE-CTF作为单一催化剂时,相应的产率在相同条件下约50%,这一结果与我们先前的猜想一致。引入的三嗪环对CO2强捕获能力可以促进环加成反应的进行。与此同时,当使用KI作助催化剂,CE-CTF其催化性能可以获得大幅度改进,在相同条件下获得了完全转化,产物选择性超过99%。上述结果证明弱碱性的三嗪环和亲核的碘离子之间的相互协同催化效应是促进环氧化物开环和CO2分子活化进而提高催化活性的关键。
当升高反应温度至120 ℃,只需8 h可完全转化,降低温度到80 ℃后反应8 h,也能获得较好的产率。为了证实开发的冠醚型CTFs的高效性,与其他已报道的催化体系的实验结果进行比较,CE-POP-2与KI的二元催化体系,在100 ℃和1.0 MPa的CO2反应条件下,反应12 h后产率达70%,这可能是由于该催化体系中缺少三嗪结构。而Poly18C6与KI的二元催化体系,在相同条件下3 h 产率达90%,这可能是由于该催化体系具有一定的均相特征,Poly18C6的线形结构可能会导致KI的浸出和部分降解。因此,KI/CE-CTF的设计方法不仅可以解决KI在有机溶剂中溶解度低的问题,而且将三嗪单元引入主体骨架中,从而实现催化性能的显著提高。
2.8 催化剂动力学及稳定性
KI/CE-CTF的动力学曲线和循环性能测试如图7所示。由图7的动力学曲线可以看出,随着反应时间的增加,目标产物的产率逐渐增大,基本符合一级反应动力学特征。因此在该条件下进一步测试了该催化材料的循环使用性。由于CE-CTF具有明显的异质性的特点,通过简单离心或过滤操作即可实现催化剂的回收。由图7的循环性能测试可以看出,在100 ℃和1.0 MPa条件下,反应12 h,环状碳酸酯产率保持约65%。表明CE-CTF催化剂循环使用5次后,催化活性未见明显下降,展现出良好的循环稳定性。CE-CTF的催化活性在连续使用后活性有小幅度下降,在其他类似的催化体系中也能观察到这种情况[21]。
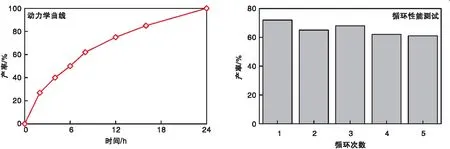
图7 KI/CE-CTF的动力学曲线和循环性能测试Figure 7 Dynamic curve and cyclic performance test of KI/CE-CTF
2.9 催化剂底物拓展性
为了评价该双组分催化系统KI/CE-CTF的底物拓展性,将多种环氧化物作为底物转化为对应的环状碳酸酯,在环氧化物5.0 mmol、KI/CE-CTF为20 mg、CO2压力1.0 MPa和反应温度100 ℃条件下,CO2与各种环氧化合物在KI/CE-CTF催化下的加成反应结果如表2所示。环加成反应式为:
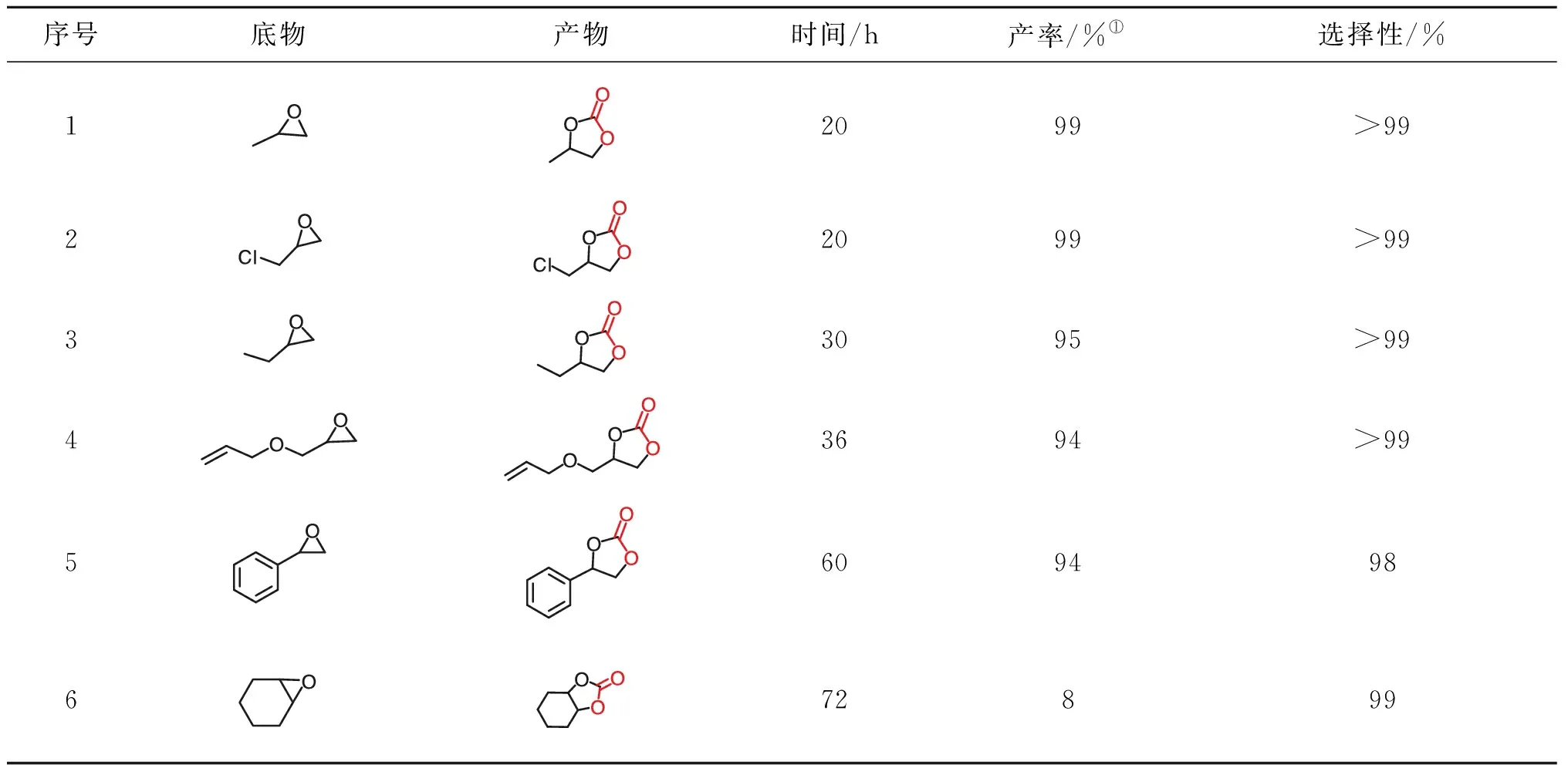
表2 CO2与各种环氧化合物在KI/CE-CTF催化下的加成反应结果Table 2 Results of addition reaction of CO2 with various epoxides catalyzed by KI/CE-CTF

由表2可以看出,KI/CE-CTF二元催化体系对大部分末端环氧底物都具有良好的催化活性和高选择性(>99%)。当使用环氧丙烷和环氧氯丙烷作为底物时,反应20 h就可以完全转化,而1,2-环氧丁烷需要进一步延长反应时间至30 h,产物产率达95%。这是因为与环氧丙烷相比随着烷烃支链长度的增加,其转化难度增加。在相同条件下,含有碳碳双键的烯丙基缩水甘油醚作为底物时,反应36 h,环状碳酸酯产率达94%,展现出该催化体系具有一定的官能团适用性。对于相对惰性的芳香类环氧化物(如氧化苯乙烯),要获得相对较高的转化率,则需要进一步延长反应时间至60 h。对于内环化合物氧化环己烷,反应72 h,产率仅为8%,原因可能是空间位阻过大,阻碍了环氧化物的开环,导致其催化转化性能差,这一结果与绝大多数相似的催化体系一致。
2.10 反应机理探究
KI/CE-CTF催化CO2与环氧化物环加成反应的可能机理如图8所示。由图8可以看出,一方面,环氧化物和三嗪环之间的相互作用会增强环氧化物的活性,从而增加附着在碳原子上的正电荷密度[27]。与此同时,三嗪环上的氮原子与CO2结合形成氨基甲酸酯,氨基甲酸酯的氧负离子以亲核试剂的方式攻击环氧化物受阻较小的一侧,导致环氧化物开环和形成碳酸酯中间体,最后,氧负离子和亲电碳原子相互作用,通过分子内环闭合生成环状碳酸酯。另一方面,活化的亲核碘离子也可能直接进攻环氧化物上面阻止相对较小的一侧,促使环氧化物开环;在富氮三嗪环的作用下,CO2被富集在活性位点附近,CO2分子插进高活性氧负离子中间体,从而形成碳酸盐过渡物种,最后发生分子内闭环生成相应的环状碳酸酯。

图8 KI/CE-CTF催化CO2与环氧化物环加成反应的可能机理Figure 8 Possible mechanism of the catalytic cycloaddition reaction between CO2 and epoxides over KI/CE-CTF
3 结 论
通过醛脒缩合反应在温和条件下合成了一种冠醚型CTFs。依次采用FT-IR、固体核磁、XPS、TEM、N2吸附-脱附和热重对CE-CTF的结构和形貌等进行了系统表征与分析。利用三嗪基团对CO2的强相互作用以及冠醚大环对钾离子的螯合作用,当使用KI作为助催化剂时,所获得的CE-CTF催化材料在温和条件下能够催化CO2与环氧化物的环加成反应,并表现出优异的催化活性。同时,该催化剂具有良好的催化循环稳定性和热稳定性,并对大部分末端环氧化物都具有良好的催化活性,表现出杰出的底物适用性。本研究为CO2环加成多相催化剂的设计与开发提供了一条清晰而完整的思路。
猜你喜欢
科学大众(2023年17期)2023-10-26
无机化学学报(2021年11期)2021-11-18
小天使·二年级语数英综合(2021年5期)2021-07-11
化工学报(2021年6期)2021-06-30
高等学校化学学报(2020年3期)2020-03-12
中学化学(2017年2期)2017-04-01
咸阳师范学院学报(2016年6期)2017-01-15
合成化学(2015年1期)2016-01-17
浙江化工(2015年9期)2015-11-25
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
