
朗格汉斯细胞组织细胞增生症临床特点及其BRAF V600E 基因突变与预后的相关性*
2024-04-18 08:26朱丽华金皎高勤吴莎莎黄璟
贵州医科大学学报 2024年2期
朱丽华, 金皎*, 高勤, 吴莎莎, 黄璟
(1.贵州医科大学附属医院 儿科血液专科, 贵州 贵阳 550004; 2.贵州医科大学附属医院 病理科, 贵州 贵阳 550004)
朗格汉斯细胞组织细胞增生症(langerhans cell histiocytosis,LCH)是一种罕见的以朗格汉斯细胞增生为特征的一组病变,病因尚未明确[1]。LCH可能侵及骨骼、皮肤、淋巴结或实质器官,如肝、脾、骨髓,临床以单系统病灶发病多见,常见于骨骼系统。该病临床表现差异极大,轻者仅累及骨骼、皮肤,重者可累及多器官并造成重要脏器功能损害,患者预后较差,其发病率与年龄呈负相关,好发于儿童(约占70%),随年龄增长,发病率逐渐降低,各个年龄段均可发病[2]。儿童发病率约为 (3~5)/100 万[3],成人相对较少,发病率约为(1~2)/100 万[4]。LCH起病症状较隐匿,临床表现复杂多样,具有高度异质性,临床易漏诊或误诊。研究发现其存在着多种基因突变,包括BRAF V600E、ARAF 及 MAP2K1 基因等,其中BRAF V600E基因突变对于疾病的预后判断依旧存在着争议,仍是一个尚未解决的难题。本研究对收治的17例LCH患者的临床特点、组织病理学特点、治疗结局、BRAF V600E基因突变情况进行整理分析,以提高对LCH的认识,同时统计分析LCH中BRAF V600E基因的突变率及其对于患者预后的影响,从而进一步分析它的临床意义。
1 对象与方法
1.1 研究对象
选取2014年2月—2020年6月收治的17例LCH患者作为研究对象,纳入标准:(1)符合国际组织细胞协会2009年的LCH评估与治疗指南的诊断标准[5];(2)有本院活检组织病理确诊的病例;(3)进行BRAF V600E基因突变情况检查;(4)资料信息完整;(5)治疗后随访。排除标准:(1)未达确诊标准或无本院活检组织病理确诊的病例;(2)未进行BRAF V600E基因突变情况检查;(3)资料信息不全;(4)治疗后未随访。
1.2 研究方法
1.2.1临床特点分析 记录LCH患者的首发病变部位(皮肤、淋巴结、肝脾、肺部、耳、骨、中枢神经系统)及各病变部位的具体表现。
1.2.2组织学检查 取患者首发病变部位组织,其中骨组织预先经脱钙处理,采用苏木精-伊红染色法(hematoxylin-eosin staining,HE )进行组织学检查,切片(厚度5 μm ,长度0.5 cm)平铺于38 ℃纯水,待伸展完全后捞出,放入60 ℃烘箱烤片5 h,经常规脱蜡、水化,苏木素染色3~5 min、水洗5 min,盐酸酒精分化数秒后水洗5 min,伊红染色1 min、水洗5 min,经70%、80%、90%、100%梯度乙醇脱水各1次、每次10 s,浸入二甲苯3 min、中性树胶封片。
1.2.3基因检测与分组 通过数字聚合酶链式反应(polymerase chain reaction,PCR)扩增和一代测序的方法测定患者首发病变部位组织标本中的BRAF V600E 基因,提示BRAF V600E 阳性,则判为阳性,完善检测,无阳性结果则判为阴性;根据LCH临床分型将17例患者分为单系统病变(singlesystem Langerhans cell histiocytosis,SS-LCH)组和多系统病变(multi-system Langerhans cell histiocytosis,MS-LCH)组以及根据年龄分为未成年组和成年组。
1.2.4治疗与预后 根据LCH患者病变范围及受累部位,实施具体治疗手段,包括手术切除、化学药物治疗及放射治疗;统计治疗手段,通过随访记录预后情况。
1.3 统计学分析
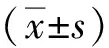
2 结果
2.1 临床特点分析
17例患者中,病灶首发于骨骼13例(76.47%),累及部位包括额骨、顶骨、枕骨及颞骨等部位,X线、CT或MR上可表现为局限性或多发性骨质破坏伴软组织肿块形成、密度减低,头颅骨可表现为骨质缺损,椎体损害表现为椎体变扁、骨质破坏并椎体压缩;首发于皮肤者2例(11.76%),均为婴儿期患儿,受累部位以颜面部、躯干部多见,表现为迁延不愈、新旧不一的淡红色或紫红色丘疹、湿疹样皮疹,部分结痂,皮疹高出皮肤表面,中间凹陷,压之不褪色,可伴少许鳞屑、脂溢性皮炎,皮疹消退后可遗留色素脱失斑等,外用药物治疗效果欠佳;首发于淋巴结患者2例(11.76%),多数大小在0.5~5.0 cm,质中,无压痛,部分可融合成团,与周围组织粘连,多累及双侧颌下、颈部、腋窝、腹股沟处等淋巴结;首发于肝脾部位患者4例(23.53%),其中合并肝、脾受累3例(17.65%),多表现为肝脏肿大、最大肋下5 cm、CT上表现为肝脏体积增大伴多发小结节状低密度影、肝脏囊肿等,单独脾脏受累1例(5.88%),表现为脾脏肿大、最大肋下5 cm、无明显脾功能亢进症状;首发于肺部患者1例(5.88%),表现为咳嗽、咳痰、呼吸困难;合并耳受累1例(5.88%),表现为耳朵疼痛,伴听力下降,反复耳朵流脓,抗感染治疗效果欠佳;首发于中枢神经系统合并有尿崩症者1例(5.88%),垂体MR可见垂体柄结节影。本次收集17例病例中,2例首诊于皮肤科,1例首诊于耳鼻喉科,5例首诊于儿科,余9例首诊于骨科,首个症状出现至明确诊断时间为7 d至3年不等。由此可见,本病累及器官多、临床表现多种多样是导致误诊及延长诊断时间的主要原因,需结合临床表现、病理学特点、免疫组化结果确诊。见表1。

表1 17例患者的临床特点Tab.1 Clinical characteristics of 17 cases with LCH
2.2 组织学检查
结果显示,病变组织主要由朗格汉斯细胞及嗜酸性粒细胞组成,可伴数量不等的淋巴细胞、泡沫样组织细胞,局灶区域可见肉芽肿形成及多核巨细胞,朗格汉斯细胞中等偏大,核呈卵圆形、短梭形或椭圆形,可见核折叠、核沟(图1);其中朗格汉斯细胞阳性表达白细胞分化抗原1a(cluster of differentiation 1a,CD1a) 100%(17/17),钙结合蛋白(calcium-binding protein,S-100) 100%(17/17),细胞核增值相关抗原Ki-67(nuclear proliferation associated antigen Ki-67) 88%(15/17),白细胞分化抗原68(cluster of differentiation 68,CD68)82%(14/17),细胞因子(cytokine,CK)、白细胞分化抗原3(cluster of differentiation 3,CD3)、白细胞分化抗原20(cluster of differentiation 20,CD20)、上皮膜抗原(epithelial membrane antigen,EMA)均阴性。
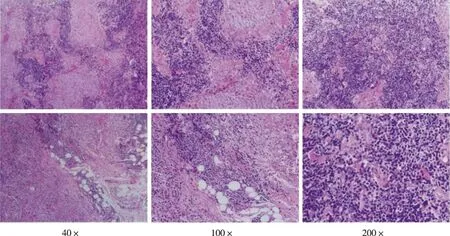
图1 部分患者病变部位的朗格汉斯细胞HE染色Fig.1 HE staining of Langerhans cells in some patients with LCH
2.3 基因检测与分组
结果表明,7例检出BRAF V600E 基因突变,总阳性率41.18%;其中SS-LCH组13例,BRAF V600E 基因突变7例,阳性率53.85%,MS-LCH组未发生BRAF V600E基因突变。未成年人组12例,其中BRAF V600E 基因突变5例,阳性率41.67%,在3岁以下低龄儿童病变中BRAF V600E基因突变率是3岁以上的2倍,成年人组共5例,其中BRAF V600E 基因突变2例,阳性率40.00%,这17例患者的BRAF V600E基因分别在多系统与单系统、未成年人与成年人组间比较中,差异均无统计学意义(P>0.05)。见表2、表3。

表2 SS-LCH与MS-LCH组BRAF V600E基因突变阳性率分析Tab.2 Analysis of positive rate of BRAF V600E gene mutation between SS-LCH group and MS-LCH group
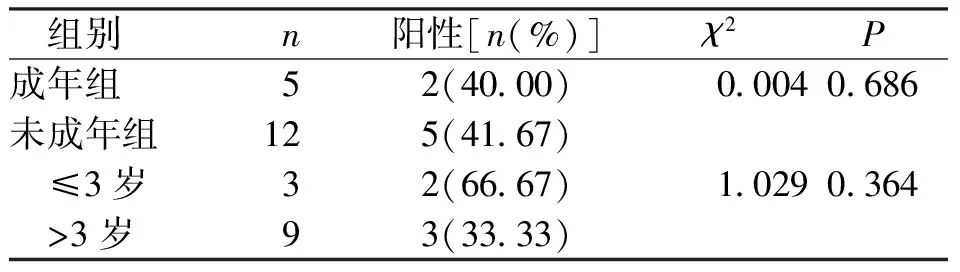
表3 成年组与未成年组BRAF V600E基因突变阳性率分析Tab.3 Analysis of BRAF V600E gene mutation positive rate between the adult group and juvenile group
2.4 治疗与预后
结果表明,单纯化疗7例、单纯手术治疗5例、手术+化疗4例、手术+化疗+放疗1例;其中以化疗为主的全身治疗最多,占 41.18%,其次为单纯手术治疗,占29.41%,手术+化疗占23.53%,手术+化疗+放疗占5.88%;化疗方案包括VP化疗方案(长春新碱+泼尼松)、CHOP化疗方案(环磷酰胺+多柔比星+长春新碱+泼尼松) 、COP化疗方案(环磷酰胺+长春地辛+泼尼松)、LCH-Ⅲ化疗方案(长春花碱+泼尼松)、DAL-HX83/90化疗方案(泼尼松+长春地辛+依托泊苷+甲氨喋呤+巯嘌呤)、CCAP( 环磷酰胺+阿糖胞苷+泼尼松)、SCMC-LCH-2008(上海儿童医学中心LHC方案),均未采用维罗非尼靶向治疗。随访 5~81 个月,中位随访时间58个月,其中SS-LCH组均治愈,MS-LCH组中1例患者随访8个月后死亡,3例患者分别随访41个月、48个月、52个月时出现复发,且均无BRAF V600E基因突变。SS-LCH组预后较好,MS-LCH组预后较差,但差异无统计学意义(P>0.05);复发患者现仍继续治疗中,共16例患者生存至今,其中复发及死亡患者均为未成年组患儿,受累器官较多,其余患者均治愈,目前均未复发,7例BRAF V600E基因突变患者治疗效果尚可,均已治愈,提示BRAF V600E 基因突变对生存的影响差异无统计学差异(P>0.05)。见表4。

表4 BRAF V600E基因突变阳性与阴性患者预后分析(例)Tab.4 Prognosis analysis of BRAF V600E gene mutation positive and negative cases
3 讨论
LCH是一种以朗格汉斯细胞组织细胞异常增殖为病理特征的罕见疾病。由于LCH特有的形态学表现(组织细胞形态上含有大细胞的组织病变),多种研究表示其与巨噬细胞、嗜酸性粒细胞、淋巴细胞、巨细胞有关[6],长期以来一直被归类为单核 - 巨噬细胞系统的疾病[7]。LCH 病变包括浸润的病理性树突状细胞、巨噬细胞、嗜酸性粒细胞和富含调节性T细胞的淋巴细胞。有研究表明树突细胞表达高水平的T细胞共刺激因子和促炎细胞因子,细胞因子与趋化因子受体构成“细胞因子风暴”[8]。近年来,研究证明大量的树突状细胞可表达多效细胞因子骨桥蛋白,促进T辅助细胞的产生,组织细胞招募单核细胞和破骨细胞活化,同时也会产生大量组织破坏性酶[9],可导致受累组织的损坏,约75%患者激活的体细胞中发现BRAF V600E(57%),提示 LCH 有肿瘤增殖的过程[10]。BRAF 突变是LCH的诊断和预后指标,也是 BRAF V600E 特异性抑制剂靶向治疗的重要指标[11]。LCH尚缺乏标准的治疗方案,化疗方案多种多样,包括单用泼尼松、泼尼松和长春新碱联合用药,外科治疗包括皮肤病变局部治疗、骨病灶刮除等。近年来,BRAF V600E突变的发现改变了研究者对LCH发病机制的认识以及对治疗的选择[10]。程晓烨等[12]经过对中国50例LCH患者的研究发现BRAF V600E突变率可高达58%,研究表明BRAF V600E突变激活下游MEK/ERK通路,从而促进肿瘤的发生与发展,在无BRAF V600E突变的LCH患者中,还有部分患者具有MAP2K1的突变。BRAF抑制剂维罗非尼,在治疗BRAF V600E阳性的LCH患者中取得了不错的疗效,有效率达42%[13]。遗憾的是,在本研究中并未进行MAP2K1基因的检测及未针对BRAF V600E阳性的LCH患者使用维罗非尼治疗。
LCH病变累及单系统到多系统,可受累全身骨骼,头颈部受累较为常见,常见受累骨骼包括颌面骨及颞骨[14];累及耳颞部时,可出现耳颞部疼痛、肿胀、流脓,甚至鼓膜穿孔、耳聋等症状;累及颅底时,可出现头痛、脑脊液漏;如累及垂体时可出现尿崩症[15]。患者经常首诊于骨科、耳鼻喉科,所以临床上遇到儿童患病就诊时要多加注意,如伴有颅骨缺损、头颅包块、皮疹、外耳道流脓等症状时应考虑LCH的可能。对头颅包块的定位可通过头颅CT及MR的检查进行,LCH颅骨病变在CT及MR可有特征性表现[16],典型的CT表现为大块溶骨性破坏,具有特征性“地图状”外观,病变不规则强化。另外,LCH所引起的中枢神经系统病变损伤常局限于下丘脑-垂体 ,因此当患者出现多饮、多尿等尿崩症症状时应首选MR检查以明确脑内病变[17]。影像学检查结合临床表现有助于早期诊断及评估预后[18],而至关重要的是要进行病变部位的组织病理学检查及免疫组织化学检查以便确诊。
本研究中,17例患者均进行BRAF V600E基因突变检测,基因突变率为41.18%(7/17)。Badalian-Very 等[10]在石蜡包埋的LCH组织切片上检测到57%的BRAF V600E基因突变,并通过免疫荧光染色检测BRAF V600E突变的LCH细胞中2个下游激酶 MEK/ERK 的活化。Satoh等[19]和 Kansal等[20]在 CD1a+LCH 细胞上证实了BRAF V600E基因突变的存在。多个研究小组在LCH患者中检测到BRAF V600E基因突变,阳性率为 38% ~ 69%[21],本研究中BRAF V600E基因突变阳性率与相关报道文献相符。本研究发现未成年组发生BRAF V600E 基因的突变率与成人组差异无统计学意义(P>0.05),或因病灶标本量少,且基因突变在LCH中的研究处于起步阶段所以仍有许多问题需系统且深入的研究,该结论具有一定的局限性。Allen等[22]发现BRAF V600E基因突变发生在髓系树突状细胞中会形成高风险的LCH,而突变发生在分化成熟的树突状细胞中会形成低风险的LCH,提示 BRAF基因和LCH预后是密切相关的;而且,有研究也证实BRAF V600E基因突变会导致LCH预后变差[23]。然而,本文研究未发现BRAF V600E基因突变对LCH的生存有直接影响,可能是因为参与本次研究的标本大多数是单系统性的疾病,预后本来就很好[24];或者说这个基因突变发生在分化成熟的树突状细胞中,形成了低风险的LCH,但这需要进一步的实验研究去证实;也可能是由于参与本次研究的LCH标本病变部位大部分都来源于骨(76.5%),有学者做了大标本的临床研究,发现在LCH中病变部位如果侵及到骨组织会比未侵及到骨组织的预后要好[25]。
总之,LCH 临床表现复杂多样,易误诊,确诊需要依靠组织病理学检查,不同分型组在脏器受累范围和程度指标上有明显不同,其治疗方案必须根据患者发病部位和病变范围进行相应调整[26]。LCH 病变严重程度差异大,主要侵及的部位是骨组织,且预后良好,但累及重要器官的多系统病变预后较差。LCH常见的突变基因是 BRAF V600E,其在多系统与单系统、未成年人与成年人组间比较中,差异均无统计学意义(P>0.05),但3岁以下儿童病变中BRAF V600E基因突变率有增高的趋势,未观察到BRAF V600E 基因突变对生存的有影响,但尚需扩大样本进一步研究。
猜你喜欢
家教世界·创新阅读(2022年4期)2022-05-07
情感读本·道德篇(2021年7期)2021-12-14
风流一代·经典文摘(2021年10期)2021-10-25
中华养生保健(2020年1期)2020-11-16
国际放射医学核医学杂志(2020年2期)2020-05-30
小品文选刊(2016年1期)2016-02-12
中国药业(2014年24期)2014-05-26
中国医学科学院学报(2014年6期)2014-03-11
教育与职业(2014年28期)2014-01-19
中医研究(2013年10期)2013-03-11
