
注射用头孢哌酮钠质量评价
2024-05-03 13:33李茜杨博涵李倚天张婷婷李洁刘英
中国抗生素杂志 2024年3期
李茜?杨博涵?李倚天?张婷婷?李洁?刘英
摘要:目的 對国产与进口注射用头孢哌酮钠的质量现状进行评价。方法 采用法定标准检验与探索性研究相结合,对国家药品抽检的66批次样品与调研企业收集的样品进行检验与分析。结果 法定标准检验结果显示,66批次注射用头孢哌酮钠合格率为100%。探索性研究揭示,《中国药典》2020版标准的有关物质方法专属性较差,限度较宽,难以保证安全性,建立专属性较强的有关物质方法并可同时控制聚合物杂质;部分企业的样品检出微量的遗传毒性杂质NDMA,建议企业加强对原料生产的监控;国内样品晶体聚结较严重、粒度分布不均匀,复溶时间较长,提示国产原料的结晶工艺仍有提升空间;配伍结果显示部分制剂企业说明书中推荐的配伍稀释液的合理性值得商榷;综合影响因素与加速试验结果,结合各国药典的贮藏条件,表明统一规定该品种在凉暗处或25 ℃贮藏可能存在一定的风险;包材相容性结果显示覆膜胶塞与中硼硅玻璃瓶更能提升样品的质量。结论 目前国产注射用头孢哌酮钠整体质量一般,质量标准有待提高,建议企业优化原料结晶工艺,同时选择质量更好的包材。
关键词:注射用头孢哌酮钠;质量评价;安全性;有效性;配伍;稳定性;包材相容性
中图分类号:R917文献标志码:A
Quality evaluation of cefoperazone sodium for injection
Abstract Objective This study evaluated the quality condition of domestic and imported cefoperazone sodium for injection. Methods The statutory standard methods combined with exploratory research were used to evaluate the quality of the 66 batches of samples from the national drug market and from pharmaceutical manufacturers. Results The legal test results showed that the qualified rate of 66 batches of cefoperazone sodium for injection was 100%. But exploratory research showed that the specificity of the related substance method of ChP2020 was not good; the limits were broad, so it was difficult to guarantee safety. The related substance method with strong specific properties was established, which could control polymer impurities at the same time. The samples from some pharmaceutical manufacturers detected a trace mount of genetic impurity NDMA. It was suggested that pharmaceutical manufacturers should strengthen the monitoring of raw material production. There were also some problems in domestic samples, such as crystal coalescence; the particle size distribution was not uniform; and the resolution time was long, which all indicated that the crystallization process of domestic raw materials still had much to be improved. The compatibility results showed that the rationality of the compatibility diluents recommended in the instructions of some pharmaceutical manufacturers was questionable. The results of the influence factors and the accelerated test, combined with the storage conditions of pharmacopoeia from various countries, showed that there may be certain risks associated with samples being stored in a cool, dark place at 25 oC. The results of package material compatibility showed that the quality of the sample could be improved by the coated rubber plug and the medium-borosilicate glass bottle. Conclusion The quality of domestic cefoperazone sodium for injection at present is normal. The current statutory standard needs to be improved. It is suggested that pharmaceutical manufacturers optimize the crystallization process of raw materials and choose better-quality packaging materials.
Key words Cefoperazone sodium for injection; Quality evaluation; Security; Effectiveness; Compatibility; Stability; Package compatibility
头孢哌酮钠为第三代头孢菌素,1979年由日本富山化学工业公司研制,同年将技术与销售权转让给辉瑞公司。注射用头孢哌酮钠1980年由辉瑞公司在欧洲上市,商品名为“Cefobid”,1983年上海第三制药厂与太原制药厂分别仿制成功,1985年在我国上市[1-2]。该品种主要通过抑制细菌细胞壁的合成而产生杀菌作用[3],临床上主要用于治疗上下呼吸道感染、腹腔内感染、皮肤和软组织感染等。
头孢哌酮钠仅注射用头孢哌酮钠一种剂型。查询国家药品监督管理局网站,注射用头孢哌酮钠国内生产企业67家,批准文号181个,涉及6种规格,分别为0.5、0.75、1.0、1.5、2.0与3.0 g。进口药品企业1家,批准文号2个,涉及2种规格,分别为1.0 g与2.0 g。《中国药典》2020年版(以下简称ChP2020)[4]、日本药局方第18版(简称JP18)[5]及进口注册标准JX20070031均收载注射用头孢哌酮钠。
2023年,国家药品抽检从22个省(直辖市、自治区)抽取到66批次注射用头孢哌酮钠,涉及生产企业5家(国内4家,进口1家),批准文号6个(国内5个,进口1个)。本文按ChP2020与JX20070031的注射用头孢哌酮钠质量标准进行法定标准检验,并基于质量源于设计(QbD)理念,从原料、生产工艺、包材及说明书等方面围绕安全性、有效性、配伍、稳定性、包材相容性与质量可控性等进行探索性研究,对其质量现状进行客观评价,为进一步提高产品质量与完善标准提供依据。
1 仪器与试药
1.1 仪器与试剂
Waters2695高效液相色谱仪(Waters公司);LC-30AD-QTOF LCMS-9030四级杆飞行时间质谱仪、XRD-7000型X射线衍射仪及IRP restige-21傅里叶红外变换光谱仪(Shimadzu公司);Thermo QE plus离子轨道阱高分辨质谱仪、FEI Q45扫描电子显微镜及DIONEX ISC-5000+离子色谱仪(Thermo公司);7010B GC/MS Triple Quad气相质谱联用仪、7890B气相色谱仪及1260 Infinity Ⅱ液相色谱仪(Agilent公司);Mastersizaer2000型激光粒度仪(马尔文公司);STY-1A渗透压测定仪(天大天发公司);FlexStation3酶标仪(MD公司);ECLIPSE 80i显微镜(Nikon公司);PerkinElmer NexION型电感耦合等离子体质谱仪(PE公司);MARS 6型微波消解仪(美国CEM公司);S400-K pH多参数测定仪与XPE205电子天平(Mettler公司);2100AN浊度仪(Hach公司)。
甲醇、乙腈、磷酸、甲酸、甲磺酸和乙酸乙酯均为色谱纯,水为MilliQ去离子水,其余试剂均为分析纯。
1.2 试药
标准物质:头孢哌酮(批号130420-201105),头孢哌酮杂质A(批号130420-202006),1-甲基-5-巯基四氮唑(即头孢哌酮杂质C,批号130617-201702),7-氨基头孢烷酸(即头孢哌酮杂质E,批号130538-200902)及N-亚硝基二甲胺(批号510166-202003)均来源于中国食品药品检定研究院;头孢哌酮杂质B(批号PITBPT-B-EP-YT0704-01),头孢哌酮杂质D(批号PITBPT-D-EP-20191024-01),头孢哌酮杂质F(批号PITBPT-F-EP-YN1206-01),头孢哌酮杂质G(批号PITBPT-G-20180727-01),头孢哌酮杂质547(批号PITBPT-TSD-20200516-01),双母核二聚体杂质(批號PITBPT-DP-20200619-01),7-TMCA杂质(批号P17-TMCA-20190401-01)及头孢哌酮二聚体杂质(批号PITBPT-Dimer-yn1122-01)均来源于广州牌牌生物科技有限公司;氧哌嗪酸杂质(批号230100022)来源于C制药有限公司;氧化化合物杂质(批号XH19-2296),开环化合物杂质(批号XH19-2297)均来源于Hi-Chemical.ING;N-亚硝基二乙胺(批号F0032407)来源于北京Mahage生物技术公司;钠元素标准溶液(批号22C019-7),钾元素标准溶液(批号228044-2)及混合元素标准溶液(批号23D60460与23D61181)均来源于国家有色金属及电子材料分析测试中心;六甲基环三硅氧烷(批号C12873477),八甲基环四硅氧烷(批号C10904254),十甲基环五硅氧烷(批号C14951256)及十二甲基环己硅氧烷(批号C14751549)均来源于上海麦克林生化科技有限公司;抗氧剂2,6-叔丁基-4-甲基-苯酚(批号BCCC0032)来源于Aldrich公司;抗氧剂168(批号L990U58),抗氧剂330(批号LF70T68),抗氧剂1010(批号LK20V115)及抗氧剂1076(批号LRB0U122)均来源于河北百灵威超精细材料有限公司;抗氧剂3114(批号VQKUH-NY)来源于东京化成工业株式会社。
动物:家兔,雌雄各半,体重1.8~2.2 kg,购自北京富龙腾飞实验动物研究院有限公司。
样品:2023年国家药品抽检样品注射用头孢哌酮钠66批次,涉及5家生产企业(分别为4家国内企业A、D、E、F与1家进口企业);调研收集7批次头孢哌酮钠原料,涉及3家生产企业(分别为A、B、C企业)。
2 试验方法
2.1 法定标准检验
共66批次样品,其中64批次国内样品执行ChP2020注射用头孢哌酮钠质量标准,主要项目包括性状、鉴别、溶液的澄清度、吸光度、酸度、有关物质、头孢哌酮聚合物、水分、不溶性微粒、细菌内毒素、无菌、可见异物、装量差异与含量测定。2批次进口制剂执行JX20070031标准,与ChP2020标准相比,增加了比旋度、溶液的澄清度与颜色项目。
2.2 探索性研究
2.2.1 安全性
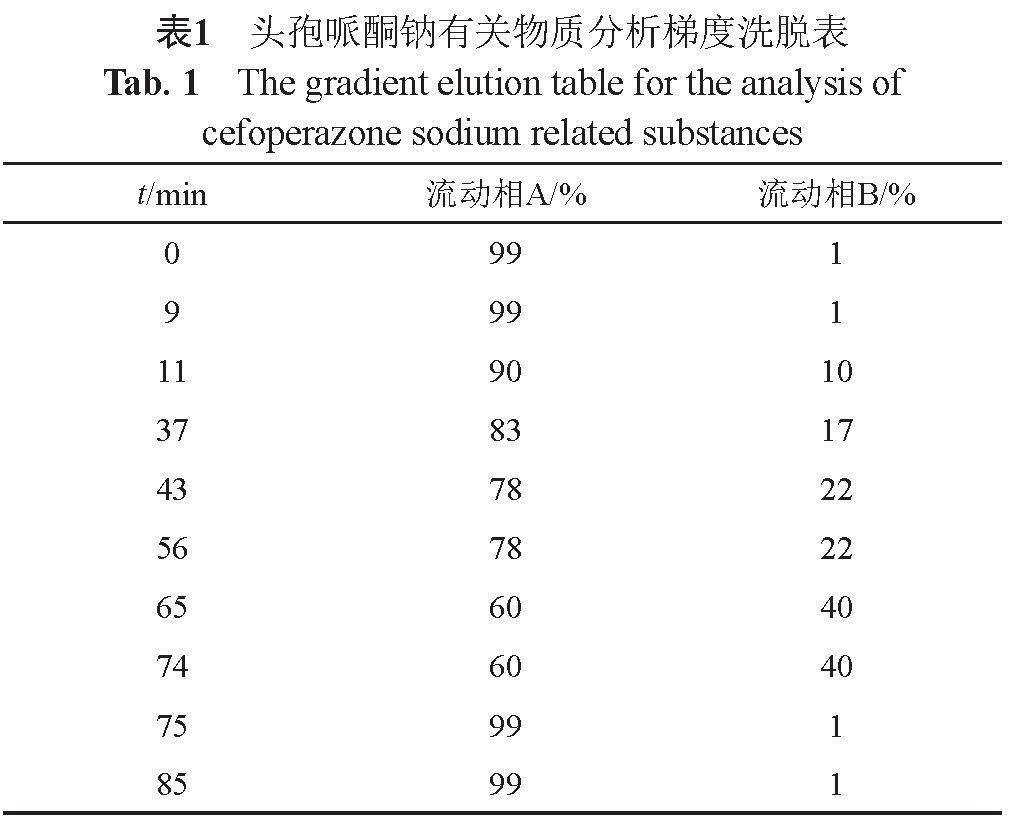
(1)有关物质 建立新的有关物质HPLC分析方法:采用Waters Xbridge C18色谱柱(150 mm×4.6 mm,5 ?m);流动相A为1.76 g磷酸氢二铵溶于1 L水中,用磷酸调节pH值至2.5,流动相B为甲醇,梯度洗脱,洗脱程序见表1,流速为1.0 mL/min;柱温30 ℃,检测波长254 nm,进样体积20 μL。
采用新建方法测定7批次原料与66批次制剂的有关物质,对原料、国内制剂与进口制剂的杂质谱进行比较分析。
探索性研究中分别采用高效凝胶色谱法与新建的有关物质方法测定66批次样品中的聚合物含量,并与葡聚糖凝胶G10法进行比较分析。
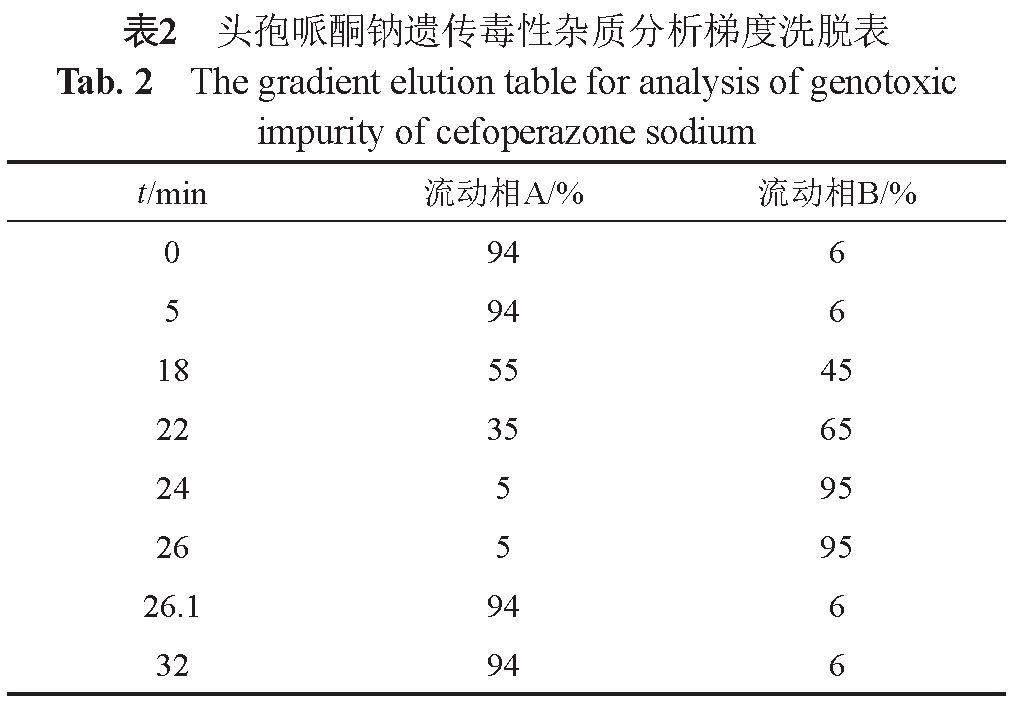
(2)遗传毒性杂质 建立UPLC-MS/MS测定注射用头孢哌酮钠7批次原料、66批次制剂与影响因素试验样品中的N-亚硝基二甲胺(NDMA)与N-亚硝基二乙胺(NDEA)。色谱条件:采用GL Sciences InertsilTM ODS-3(100 mm×3.0 mm, 3 μm)色谱柱;流动相A为含0.1%甲酸的水溶液,流动相B为含0.1%甲酸的甲醇溶液,梯度程序见表2,流速为0.3 mL/min;柱温35 ℃;进样体积10 μL。质谱条件:采用电喷雾离子源(ESI源),正离子模式检测,喷雾电压3.5 kV,离子传输管温度350 ℃,辅助气温度350 ℃,辅助气流量10 mL/min,鞘气流量40 mL/min,扫描模式为选择离子监测(SIM),质谱采集时间为3.0~6.0与13.0~16.0 min。目标离子精确质量数m/z(+H)为75.05538(NDMA)与103.08674(NDEA)。
2.2.2 有效性
(1)复溶时间 66批次样品按标示量每瓶分别加注射用水、0.9%氯化钠注射液、5%葡萄糖注射液、5%葡萄糖-0.9%氯化钠注射液各适量,制成浓度分别为含头孢哌酮0.2 g/mL的溶液,以2次/s的频率反复倒转样品并观察其在不同稀释液中的复溶情况。
(2)粒度与粒度分布 采用激光粒度仪对66批次样品进行测定。测定方式为干法,振动进样速度为60%,分散气压为0.8 bar。
(3)晶型与晶癖 采用粉末X射线衍射对原料与制剂分别进行晶型测定,方法为:阳极靶材料CuKαl,管压40 kV,管流30 mA,扫描范围(2θ)5°~60°,扫描速率为2°/min,扫描间隔0.01°。采用扫描电镜对原料与制剂分别进行晶癖测定,方法为:高真空模式,加速电压10.0 kV,电流10.1 μA,工作距离12.9 mm,放大倍数10000倍。
(4)钠离子含量与成盐率 采用离子色谱-抑制型电导检测器测定样品中的钠离子含量。色谱柱为DionexIonPac CS12A(4.0 mm×250 mm);检测器为电导检测器,抑制器为ERS 500;淋洗液为8 mmol/L甲磺酸溶液,流速为1.0 mL/min;柱温30 ℃;进样体积10 μL。成盐率% =(钠离子含量/22.99)/(头孢哌酮含量/645.68)。
2.2.3 配伍
(1)配伍稳定性 5家企业的样品分别用0.9%氯化钠注射液、5%葡萄糖注射液、5%葡萄糖-0.9%氯化钠注射液溶解并稀释成浓度均为25 mg/mL的溶液,在20 ℃下放置,以溶液的澄清度、不溶性微粒及有關物质为指标,考察样品24 h的配伍稳定性。
(2)渗透压摩尔浓度 根据各企业说明书的用法用量,D、E与F企业的样品均采用5%葡萄糖-0.9%氯化钠注射液作为配伍稀释液,A企业制剂与进口制剂均采用5%葡萄糖注射液或0.9%氯化钠注射液作为配伍稀释液,采用冰点下降法测定66批次样品的渗透压摩尔浓度。
(3)体外溶血试验 取家兔心脏血离心沉淀的红细胞混悬液,通过红细胞溶血(RBC)试验测定溶血率,考察注射用头孢哌酮钠与0.9%氯化钠注射液、5%葡萄糖注射液、5%葡萄糖-0.9%氯化钠注射液配伍后的体外溶血情况,评价样品与上述溶液配伍后的安全性。
2.2.4 稳定性
(1)影响因素试验 以性状、有关物质、酸度、溶液的澄清度、溶液的颜色、聚合物、水分、可见异物及含量为指标,考察高温(60 ℃)、高湿(相对湿度92.5%、25 ℃)与光照(4500 Lx)对注射用头孢哌酮钠质量的影响。
(2)加速试验 以性状、有关物质、酸度、溶液的澄清度、溶液的颜色、聚合物、水分、可见异物及含量为指标,考察注射用头孢哌酮钠在加速试验下(温度40 ℃、相对湿度75%)质量的变化。
(3)贮藏 通过稳定性试验的结果,结合企业提供的长期试验结果,再参考各国药典中该品种的贮藏条件,分析注射用头孢哌酮钠更合适的贮藏条件。
2.2.5 包材相容性
(1)相容性试验样品 5家企业的制剂在温度40 ℃、相对湿度75%条件下分别正置、倒置1个月和3个月。
(2)试验方法 首先采用GC-MS法测定胶塞中的挥发性可提取物,预测潜在的目标浸出物;新建GC法与HPLC-UV法分别测定相容性试验样品中的浸出物含量(即硅氧烷类与抗氧剂类);再对相容性试验样品进行性状、酸度、有关物质、可见异物及含量等常规项目考察;接着采用ICP-MS法测定相容性样品中的15种元素迁移量;最后采用亚甲蓝法考察样品对玻璃瓶的表面侵蚀情况。评价不同包材对注射用头孢哌酮钠质量的影响。
3 结果与讨论
3.1 法定标准检验
按法定标准检验,66批次样品均符合规定,合格率100%。对与产品质量密切相关的主要检验项目如酸度、有关物质、头孢哌酮聚合物、水分及含量测定结果进行统计分析。
(1)酸度 66批次样品pH值为5.0~5.3,均值为5.1,企业间pH值差异较小。
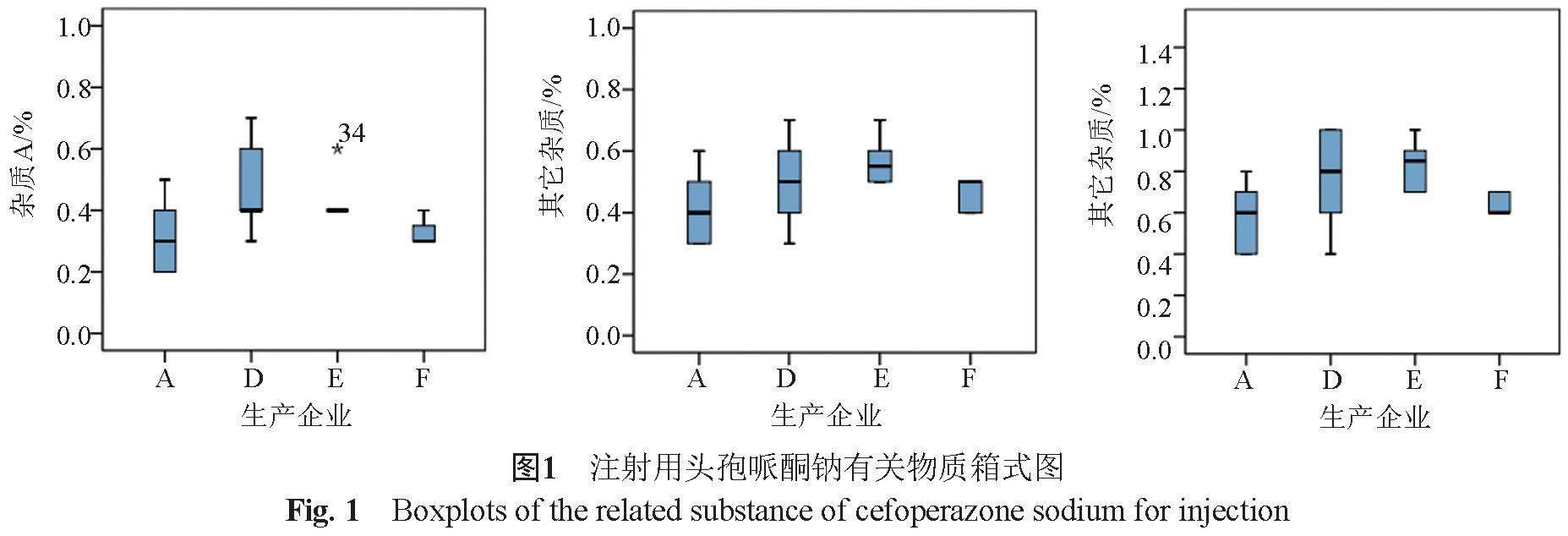
(2)有关物质 66批次样品杂质A为0.2%~0.7%,其他单杂为0.3%~0.7%,其他总杂为0.4%~1.0%;进口制剂的其他单杂与其他总杂的含量均较国内样品低。利用箱式图比较批次大于6的国产企业样品(图1):D企业样品杂质A、其他单杂与其他总杂均离散较大;E企业样品杂质A出现离群值,其可能原因为该批次样品生产日期较早,贮藏过程中降解导致了杂质A的增加。
(3)头孢哌酮聚合物 66批次样品的聚合物含量为0.03%~0.1%,均值为0.06%,远低于标准规定限值0.8%。企业间比较,进口制剂的聚合物结果最高,为0.1%,其可能原因为测定方法的不同所致。国内样品执行ChP2020标准,进口制剂执行JX20070031标准,二者均采用葡聚糖凝胶G10法,但流动相的浓度与流速均不同。采用ChP2020的方法测2批次进口制剂,结果均为0.05%,ChP2020方法的测定结果偏低。
(4)水分 66批次样品的水分含量为3.3%~4.0%,均值为3.7%。D企业样品水分离散程度较大,为3.3%~3.9%。不同企業的产品水分有差异,A企业样品均值最高,为3.9%,其可能原因为注射用头孢哌酮钠具有引湿性,不同企业原料引湿性的差异及制剂生产过程的差异可能导致成品中水分含量的不同。
(5)含量测定 按无水物计算,66批次样品的含量为91.6%~94.7%,均值为92.9%;各企业间的结果较接近,D企业样品离散程度最大,有2个离群值,分析可能与该企业样品水分离散程度较大有关。按平均装量计算,66批次样品含量为97.0%~101.6%,均值为99.3%;各企业间的结果稍有差异,E企业样品含量稍低,均值为97.7%,可能与该企业样品的平均装量稍低有关(图2)。
3.2 探索性研究
3.2.1 安全性
(1)有关物质 头孢哌酮钠稳定性较差,其在生产与贮藏过程中有可能产生较多杂质[6]。BP2023[7]与EP11.0[8]头孢哌酮钠标准中收载了6个已知杂质,分别为杂质A、B、C、D、E与F。调研收集到了其他8个已知杂质,分别为杂质G、7-TMCA杂质、氧哌嗪酸杂质、杂质547、开环化合物、氧化化合物、双母核二聚体杂质与头孢哌酮二聚体杂质。目前ChP2020标准与JX2007003标准均采用等度洗脱方式,专属性较差,无法完全分离上述杂质。限度方面仅控制了杂质A、其他单杂与其他总杂(JX2007003控制总杂)。新建的有关物质方法可分离英国药典2023年版(以下简称BP2023)与欧洲药典11.0版(以下简称EP11.0)中收载的6个已知杂质、调研收集的8个已知杂质及样品中的未知杂质,方法专属性更好(图3~4)。
采用新方法测定66批次抽检样品与调研收集的7批次原料,结果见表3,与法定标准的有关物质方法比较,新建方法检出的杂质个数更多,其他总杂含量更高,同时分析时间较短。新建方法中,杂质A按外标法计算,杂质B、杂质C与杂质E的校正因子分别为0.75、0.44与0.70,其余杂质按不加校正因子的主成分自身对照法计算。
对新建有关物质方法的测定结果进行分析,可见不同企业的原料杂质个数与杂质含量均有差异,C企业原料的杂质个数最少、杂质含量最低。制剂生产工艺对杂质的产生基本无影响;贮藏过程可导致制剂杂质增加。同一原料来源的制剂杂质个数基本相同但杂质含量有差异,原因为贮藏过程可导致杂质增加;不同原料来源的制剂杂质个数与杂质含量均有差异。进口制剂的原料来源于C企业,与国内制剂比较,进口制剂的杂质个数较少、杂质含量稍低。
进一步分析样品中的聚合物杂质。目前β-内酰胺类抗生素聚合物的测定方法主要有3种:葡聚糖凝胶G10法、高效凝胶色谱法与反相高效液相色谱法[9-10],该品种法定标准的聚合物测定方法均为葡聚糖凝胶G10法。采用高效凝胶色谱法测定66批次样品的聚合物,含量为0.5%~1.1%,均值为0.8%,结果远高于法定标准中葡聚糖凝胶G10的测定结果,其原因为高效凝胶色谱法测定的是头孢哌酮部分聚合物与小分子杂质的混合物,且无法检测到双母核二聚体,没有完全直接体现样品中聚合物的真实含量。
采用新建的有关物质方法测定该品种中的聚合物,根据LC-MS对各杂质结构推断结果,将图4中杂质15~杂质18作为聚合物杂质。66批次样品的聚合物含量为0.03%~0.2%,均值为0.1%。新方法中的聚合物不仅包括了葡聚糖凝胶G10系统中的聚合物,还包括了双母核二聚体杂质,因而绝大部分样品的结果高于法定标准中葡聚糖凝胶G10的测定结果。
(2)遗传毒性杂质 头孢哌酮钠的合成起始物料之一1-甲基-5-巯基-四氮唑,其由叠氮化物与异硫氰酸甲酯反应得到,叠氮化物中可能残留微量的亚硝酸钠,与异硫氰酸甲酯的合成起始物料一甲胺反应,有生成NDMA的风险。亚硝酸钠如传递到头孢哌酮钠的后续反应中,遇到溶剂N, N-二甲基乙酰胺或三乙胺,可能会形成NDEA[11]。A企业的原料及其相关制剂(A、D与E企业)均检出NDMA,结果为1.2~4.4 ppb,均低于限度10.6ppb,其余企业原料与制剂均未检出NDMA;所有样品均未检出NDEA;影响因素试验样品中NDMA与NDEA的含量均未增加;表明NDMA来源于头孢哌酮钠原料。
3.2.2 有效性
(1)复溶时间 66批次样品均在120 s内复溶。不同企业的样品复溶时间有差异,E企业样品复溶时间最长,在注射用水、0.9%氯化钠注射液、5%葡萄糖注射液、5%葡萄糖-0.9%氯化钠注射液中分别为76、105、107与104 s。进口制剂的复溶时间最短,在上述稀释液中依次为37、30、31与33 s。
(2)粒度与粒度分布 5家制剂企业的原料均采用碳酸氢钠成盐、丙酮溶液中结晶的溶媒结晶工艺,但制剂的粒径差异较大,粒度分布不均匀;而进口制剂粒径基本一致,粒度分布均匀。文献报道,粒径大小与粒度分布的均匀性通常与结晶温度与溶媒加入的速度有关,结晶温度较高,晶体生长速度较快,粒度较大。溶媒加入的速度合适且稳定,过饱和度值稳定,晶体生长速率稳定,粒度分布均匀[12]。提示A、B企业的原料结晶工艺仍有优化的空间。
(3)晶型与晶癖 3家原料与5家制剂的粉末X射线衍射图谱基本一致,表明均为同一晶型。扫描电镜结果显示,国内制剂与其相对应的A、B企业原料均为颗粒和短条状结晶,晶体聚结严重;进口制剂与其对应的C企业原料均为颗粒和长条状结晶,晶体聚结较轻(图5~6)。表明A、B企业的原料结晶工艺与C企业有差异。
结合复溶时间、粒度与粒度分布结果,提示国内制剂晶体聚结较严重,粒度分布不均匀,复溶时间较长。进口制剂晶体聚结较轻,粒度分布均匀,复溶时间较短。
(4)钠离子含量与成盐率 66批次样品的钠离子含量为3.2%~3.4%,成盐率为97.6%~104.8%。5家制剂的结果基本一致,均能完全成盐。
3.2.3 配伍
注射用头孢哌酮钠的给药途径主要为静脉注射,其配伍后的安全性值得关注[13]。不同企业的说明书规定的配伍稀释液不同,D企业与E企业均采用葡萄糖氯化钠注射液作为配伍稀释液;F企业说明书规定的稀释液种类较多,但也采用了葡萄糖氯化钠注射液;A企业与进口制剂均采用葡萄糖或氯化钠注射液。
(1)配伍稳定性 5家制剂分别同0.9%氯化钠注射液、5%葡萄糖注射液及0.9%氯化钠5%葡萄糖注射液配伍后室温下放置24 h,溶液均澄清、不溶性微粒均与有关物质均符合规定,表明24 h配伍稳定性良好。
(2)渗透压摩尔浓度 按说明书的规定,D、E与F企业的样品采用5%葡萄糖-0.9%氯化钠注射液作为配伍稀释液,配伍后渗透压摩尔浓度均值为597 mOsmol/kg;A企业制剂与进口制剂均采用葡萄糖或氯化钠注射液作为配伍稀释液,配伍后渗透压摩尔浓度接近正常人体血液的渗透压摩尔浓度(285~310 mOsmol/kg);5家制剂如采用5%葡萄糖注射液作为配伍稀释液,配伍后渗透压摩尔浓度接近正常人体血液的渗透压摩尔浓度。
(3)体外溶血试验 5家制剂分别同5%葡萄糖-0.9%氯化钠注射液配伍,均可出现红细胞凝聚与溶血现象,配伍浓度为25 mg/mL时溶血率均值为37.2%;而与0.9%氯化钠注射液、5%葡萄糖注射液配伍溶血率均较低,均值为5.5%。提示样品与5%葡萄糖-0.9%氯化钠注射液配伍存在安全隐患。D、E与F企业说明书均规定采用该溶液稀释样品,结合渗透压摩尔浓度结果和进口制剂说明书,3家企业配伍稀释液的合理性值得商榷。
3.2.4 稳定性
(1)影响因素试验 5家制剂企业的样品在加速实验中的变化趋势相同。表现为:①性状:高温5与10 d、高湿及光照条件下样品均无变化;高温30 d时样品的颜色均略微加深,但浅于标准规定的微黄色。②有关物质:高温条件下样品中杂质均增加;高温30 d时,其他总杂为4.1%~4.3%,增幅为2.8%~3.5%,超过标准规定的限度。高湿与光照条件下有关物质变化较小,其他总杂仅增加0.1%。③酸度:高温条件下样品的pH值均降低,降幅为0.3~0.5。高湿与光照下pH值基本无变化。提示高温条件下产生的杂质呈酸性,如图4中的杂质8(头孢哌酮开环内酯杂质)、杂质B与杂质G等。④溶液的澄清度:高温条件下,进口制剂溶液均澄清,但国内制剂高温30 d时溶液的澄清度均过规定的1号浊度标准液,其中F企业的样品变化最大,高温5 d时溶液的澄清度已超过限度,30 d时已接近3号浊度标准液。高湿与光照条件下5家制剂溶液的澄清度均低于限度。⑤溶液的颜色:高温条件下样品溶液的颜色均变深,高温30 d时溶液的颜色相当于黄色6号标准比色液。高湿与光照条件下样品溶液的颜色均保持不变,为介于黄色2号与3号标准比色液之间。即样品在高温条件下降解,产生了更多的有色杂质。⑥聚合物:高温条件下样品的聚合物含量均增加,增幅约为0.1%。高湿和光照条件下聚合物结果基本无变化。⑦水分:高温和光照条件下,样品的水分基本无变化;高湿条件下,随放置时间增加水分略增,增幅为0.1%~0.4%。⑧可见异物:高温、高湿和光照下样品未检出可见异物,即可见异物不受高温、高湿与强光照射的影响。⑨含量:高温条件下样品的含量均显著降低。高温30 d时,按无水物计算的含量与按平均装量计算的含量均低于法定标准规定的限度,其中F企业的样品含量降幅最大约为9.7%。高湿与光照条件下样品的含量基本无变化。综上,表明高温是影响注射用头孢哌酮钠质量的关键因素。
(2)加速试验 5家制剂企业的样品在加速实验中的变化趋势相同。表现为:①性状:样品的性状无变化,均为类白色结晶性粉末。②有关物质:有关物质均增加。加速试验下放置3个月时,其他总杂为1.5%~1.8%,增幅为0.7%~1.0%。③酸度:pH值均降低,降幅为0.2~0.5。即样品在高温条件下产生了更多的酸性杂质。④溶液的澄清度:加速试验下放置3个月时,进口制剂溶液的澄清度均为澄清,但国内制剂均超过限度,其中F企业的样品增幅最大,加速试验下放置1个月时溶液的澄清度已超过2号浊度标准液。⑤溶液的颜色:样品溶液的颜色均加深,加速试验下放置3个月时,溶液的颜色均相当于黄色4号标准比色液。即样品降解产生了更多的有色杂质。⑥聚合物:样品中聚合物的含量略增,增幅为0.02%~0.03%。⑦可見异物:可见异物不受加速试验的影响。⑧含量:样品含量均降低。加速试验下放置3个月时,按平均装量计算的含量均接近低限。
(3)贮藏 ChP2020标准的贮藏条件为“密闭,冷处保存”,但4家国内抽检企业在2007—2010年间取得了“国家药品监督管理局药品补充申请批件”,将贮藏条件由现行标准的“冷处保存”变更为“在凉暗处保存”。C企业的原料为“冷处保存”,但采用该原料生产的进口制剂为“25 ℃以下避光”保存。
杂质谱分析显示,贮藏过程中样品杂质含量增加。影响因素试验结果显示,高温条件下放置30 d时,5家企业的制剂有关物质含量均超限;含量亦均低于低限;酸度、溶液的颜色与聚合物含量的变化均较大;4家国内企业的制剂的澄清度均超限。加速试验结果显示,3个月时5家企业的制剂含量(按平均装量计算)接近低限,酸度与溶液的颜色变化较大;国内制剂溶液的澄清度均超限。企业提供的长期稳定性试验结果显示,样品放置24个月[温度(25±2) ℃、相对湿度(60%±5%)],有关物质增加1.3%~2.6%,pH值降低0.2~0.5,含量降低1.2%~4.3%。BP2023与EP11.0均规定头孢哌酮钠原料为“冷处”保存,JP18规定头孢哌酮钠原料与制剂均为“冷处”保存。综上分析,温度对该产品的质量影响最大,统一规定注射用头孢哌酮钠在“凉暗处”或“25 ℃以下避光”贮藏,可能存在一定的风险,说明书中根据具体产品的特性确定贮藏条件是较理想的方式。
3.2.5 包材相容性
注射用头孢哌酮钠为注射用无菌粉末,根据ChP2020四部通则9621《药包材通用要求指导原则》[14],其包材的风险程度为最高,同时制剂与包材也存在发生相互作用的风险。5家样品的胶塞均为未覆膜胶塞,且玻璃瓶材质不同。A、D与F企业的样品均为钠钙玻璃瓶;E企业1.0g规格的样品为低硼硅玻璃瓶,0.5 g规格为钠钙玻璃瓶;进口制剂为中硼硅玻璃瓶。参考《化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则(试行)》与《化学药品与弹性密封件相容性研究技术指导原则(试行)》[15-16],对注射用头孢哌酮钠与包材进行相容性考察,确保样品的安全性。
(1)胶塞中的挥发性可提取物 5家制剂的胶塞中均检出硅氧烷,主要为六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷及十二甲基环六硅氧烷; A、D企业样品的胶塞检出抗氧剂2,6-叔丁基-4-甲基-苯酚(BHT);提示胶塞中挥发性物质有迁移至样品中的风险。
(2)浸出物试验 相容性试验样品中,硅氧烷类化合物六甲基环三硅氧烷含量为0.3~1.6 ppm、八甲基环四硅氧烷含量为0.8~23.9 ppm、十甲基环五硅氧烷含量为0.2~8.4 ppm、十二甲基环六硅氧烷含量为0.1~1.7 ppm,其中八甲基环四硅氧烷含量最高,但均远低于限度266.7 ppm。随放置时间增加样品中的硅氧烷含量增加,倒置比正置含量高,表明未覆膜胶塞中的硅氧烷可不断迁移至样品中。抗氧剂类化合物:D、F企业的样品检出了BHT,分别为5.8~40.0 ppm与0.2~0.5 ppm;A、E企业的样品检出了抗氧剂1076,分别为2.2~4.3 ppm与1.2~1.8 ppm;均远低于规定限度;进口制剂均未检出6种抗氧剂。随放置时间的增加,样品中的抗氧剂含量均增加,倒置样品中抗氧剂的含量比正置样品高,表明未覆膜胶塞中的抗氧剂不断迁移至样品中。
(3)溶液的澄清度 相容性试验样品中,倒置3个月的样品溶液的澄清度均超限;更换为覆膜胶塞后,A、D企业的样品与进口制剂溶液的澄清度均符合规定,但E、F企业的样品仍超限;中硼硅玻璃瓶替换原样品的玻璃瓶,5家企业的制剂溶液的澄清度均符合规定。表明未覆膜胶塞、钠钙与低硼硅玻璃瓶均可影响溶液的澄清度。
采用SPSS软件对样品中的硅氧烷、抗氧剂含量与溶液的澄清度(浊度值)分别进行双变量相关分析,结果八甲基环四硅氧烷、十甲基环五硅氧烷与浊度值均呈中等相关,十二甲基硅氧烷、抗氧剂BHT与浊度值强弱相关,六甲基环三硅氧烷、抗氧剂1076与浊度值无相关性。即未覆膜胶塞对溶液的澄清度影响与胶塞中的八甲基环四硅氧烷、十甲基环五硅氧烷与抗氧剂BHT的迁移有关。
(4)常规检验 相容性试验样品中,酸度、有关物质与含量正置与倒置结果基本一致。
(5)元素迁移试验 相容性试验样品中,15种元素迁移量均远低于限值。
(6)玻璃内表面侵蚀试验 相容性试验样品的玻璃瓶中,均未出现亚甲蓝染色现象,表明注射用头孢哌酮钠对玻璃瓶不产生表面侵蚀作用。
综上,注射用头孢哌酮钠采用未覆膜胶塞、钠钙或低硼硅玻璃瓶影响溶液的澄清度。包材中的4种硅氧烷、6种抗氧剂及15种元素的迁移量均低于限值;样品对玻璃瓶表面无侵蚀作用。
4 总结
采取法定标准检验结合探索性研究对注射用头孢哌酮钠的质量现状进行了评价。按法定标准检验,66批次注射用头孢哌酮钠均符合规定。探索性研究显示,ChP2020的有关物质方法专属性较差、分析时间较长、限度较宽;ChP2020与JX20070031收载的聚合物测定方法对同批次样品的测定结果差异约50%;新建立的有关物质方法专属性较强,可同时控制样品中的聚合物杂质。此外,A企业原料及其相关制剂中检出微量的NDMA杂质,建议企业加强对原料生产的监控。国内制剂样品晶体聚结严重、粒度分布不均匀、复溶时间较长,提示国产原料的结晶工艺仍有提升的空间;配伍结果显示,D、E与F企业说明书中推荐的配伍稀释液的合理性值得商榷。综合影响因素与加速试验结果,结合各国药典中该品种的贮藏条件,统一规定注射用头孢哌酮钠在凉暗处或25 ℃贮藏可能存在一定的风险,根据具体产品的特性在說明书中明确贮藏条件是较理想的方式。包材相容性试验结果表明,采用换覆膜胶塞与中硼硅玻璃瓶可提升样品的质量。
参 考 文 献
桥本茂一. 注射用头孢哌酮的体内动态[J]. 国外医药抗生素分册, 1990, 11(6): 445-447.
徐文伟. 国产超广谱头孢菌素—注射用头孢哌酮技术鉴定会在沪召开[J]. 抗生素, 1986, 11(1): 79-80.
Elizabeth A, Funk M D, Larry J, et al. Antimicrobial activity, pharmacokinetics, adverse reaction, and therapeutic indications of cefoperazone[J]. Pharmacotherapy, 1982, 2: 185-196.
國家药典委员会. 中华人民共和国药典[S](2020年版二部). 北京: 中国医药科技出版社, 2020: 339-340.
The Japanese PharmacopeiaⅩⅧ[S]. Japanese Pharmacopeia committee, 2021: 681-682.
上官丹罡, 周星辰, 李苌清, 等. 头孢哌酮钠及其复方制剂中的有关物质分析[J]. 药物分析杂志, 2011, 31(10): 2013-2019.
British Pharmacopoeia Commission. The British Pharmacopoeia 2023[S]. London: The Stationary Office, 2023(Ⅰ): 473-474.
European Directorate for the Quality of Medicine & Health Care. The European Pharmacopoeia 11.0[S]. Strasboourg: European Directorate for the Quality of Medicines & Health Care, 2022: 2240-2242.
胡昌勤. β-内酰胺类抗生素聚合物杂质控制策略的形成与发展[J]. 中国新药杂志, 2020, 29(11): 1231-1244.
符雅楠, 李进, 尹利辉, 等. 头孢菌素类抗生素聚合物杂质研究进展[J]. 中国抗生素杂志, 2022, 47(2): 114-121.
张霁, 张英俊, 聂飚. 药物研发中基因毒性杂质的控制策略与方法探索进展[J]. 中国医药工业杂志, 2018, 49(9): 1203-1220.
李艳斌, 王永莉, 王静康. 头孢哌酮钠结晶工艺的进展[J]. 中国抗生素杂志, 2004, 29(1): 58-62.
刘建云. 头孢哌酮钠在常用输液中的稳定性及配伍综述[J]. 广州化工, 2012, 40(12): 27-40.
国家药典委员会. 中华人民共和国药典[S]. (2020年版四部). 北京: 中国医药科技出版社, 2020: 547-548.
国家药品监督管理局. 国家食品药品监督管理总局关于发布化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则(试行)的通告(2015年第40号)[EB/OL].[2015-7-28]https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20150728120001551.html.
国家药品监督管理局. 国家药品监督管理局关于发布化学药品与弹性体密封件相容性研究技术指导原则(试行)的通告(2018年第14号)[EB/OL]. [2018-4-26]. https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20180426165301393.html.
猜你喜欢
汽车工程师(2021年12期)2022-01-18
建材发展导向(2021年23期)2021-03-08
软件导刊(2016年9期)2016-11-07
中国实用医药(2016年26期)2016-11-07
求知导刊(2016年27期)2016-11-07
商业会计(2016年15期)2016-10-21
职业(2016年10期)2016-10-20
信息安全与通信保密(2016年3期)2016-08-23
上海医药(2016年3期)2016-03-23
中国实用医药(2016年6期)2016-03-17
