
注射用头孢唑肟钠有关物质测定方法的建立
2024-05-03 13:00张秉华王小亮梁亚伟席志芳牛龙青
中国抗生素杂志 2024年3期
张秉华?王小亮?梁亚伟?席志芳?牛龙青

摘要:目的 建立注射用头孢唑肟钠有关物质HPLC分析方法并对国产样品杂质情况行分析。方法 采用Agilent Zorbax SB-C18(250 mm×4.6 mm, 5 μm)色谱柱,柱温:35 ℃,流动相:以pH3.6乙酸铵溶液为流动相A,乙腈为流动相B,线性梯度洗脱:0→10 min,97%A→90 % A;10→20 min,90%A→80 % A;20→30 min,80%A;30→35 min,80%A→15 % A;35→45 min,15 % A;45→46 min,15%A→97%A,46→55 min,97%A;流速:0.8 mL/min,检测波长:254 nm,进样量:20 ?L。结果 酸、碱、氧化、高温、光照强制降解试验证明方法的专属性良好;头孢唑肟及头孢唑肟诸分别在0.75~4.5 μg/mL浓度范围内线性关系良好;头孢唑肟及10种已知杂质(杂质18、开环脱羧S-氧化物1、开环脱羧S-氧化物2、杂质KTO、杂质16、杂质6、杂质3、杂质13、二聚体及杂质12)的定量限分别为1.63、6.30、2.78、2.84、2.31、7.86、3.06、1.62、1.90、3.87和3.09 ng;各已知杂质平均回收率均在“96.5%~101.6%”之间;重复性RSD均符合要求。结论 所建立的方法专属性、准确性及耐用性均良好,可用于注射用头孢唑肟鈉有关物质的检测。
关键词:头孢唑肟钠;有关物质;高效液相;杂质检测
中图分类号:R917文献标志码:A
Establishment of an HPLC method for determination of related substance of cefazoxime sodium for injection
Abstract Objective This study constructed an HPLC method for the determination of the related substance cefazoxime sodium for injection and analyzed the impurities in domestic products. Methods The HPLC analysis was performed on an Agilent Zorbax SB-C18 column(250 mm×4.6 mm, 5μm) with the column temperature at 35 oC. The mobile phase A was the pH 3.6 ammonium acetate solution, and the mobile phase B was acetonitrile. Gradient elution was performed as follows: 0→10 min, 97%A→90%A; 10→20 min, 90%A→80%A; 20→30 min, 80%A; 30→35 min, 80%A→15%A; 35→45 min, 15% A; 45→46min, 15%A→97%A; 46→55min, 97%A. The flow rate was 0.8 mL/min. The detection wavelength was 254 nm. The injection volume was 20 ?L. Results The acid, alkali, oxidation, high temperature, and light-forced degradation tests demonstrated good specificity of the methods. The linear range of cefzoloxime and cefazoxime was found to be between 0.75~4.5 μg/mL with a good linear relationship. The quantification limits of cefotaxime and 10 of the known impurities (impurity 18, open ring decarboxylated S-oxide1, open ring decarboxylated S-oxide2, impurity KTO, cefzoloxime open ring, impurity D, impurity 3, cefzoloxime E isomer, dimer and impurity 12) were 1.63, 6.30, 2.78, 2.84, 2.31, 7.86, 3.06, 1.62, 1.90, 3.87 and 3.09 ng, respectively. The average recoveries of all known impurities were between 96.5% and 101.6%. The RSD of repeatability all met the requirements. Conclusion The method proved to be specific, accurate and rugged, which made it suitable for the related substance analysis of cefazoxime sodium for injection. It could be used for the control of cefazoxime sodium for injection in the domestic market.
Key words HPLC; Related substance; Impurity profile; Cefazoxime sodium
注射用头孢唑肟钠为头孢唑肟钠无菌原料直接分装制成的注射剂,适用于对头孢唑肟敏感的链球菌属、肺炎链球菌、流感嗜血杆菌、大肠埃希菌、克雷伯菌属、变形杆菌属、沙雷菌属等引起的感染,临床应用广泛[1]。新冠肺炎时期,临床应用亦较多[2-3]。
头孢唑肟钠由7-氨基-氢头孢烷酸(7-ANCA)与AE活性酯或DNMA活性酯合成,其杂质主要由合成工艺与降解途径引入[4]。《中国药典》2020年版注射用头孢唑肟钠采用HPLC法对其有关物质进行控制,头孢唑肟二聚体限度为不得过0.1%,其他单个杂质不得过0.5%,杂质总量不得过1.0%[5]。企业的稳定性资料表明,产品中的杂质量随储藏时间而增加;各生产企业的实际应用中发现药典方法不能准确地分离、检测工艺杂质和降解杂质;因而,一些企业申报了各自的企业标准,采用不同的有关物质检测方法控制自己产品的质量,提示《中国药典》2020 年版注射用头孢唑肟钠有关物质HPLC测定法需进一步地优化。
2023年注射用头孢唑肟钠列入国家药品抽检计划,抽验样品涵盖28个生产企业,涉及8个不同的有关物质检查方法。按照现行药品质量控制理念,参照相关文献[6-8]。本文尝试对《中国药典》2020年版注射用头孢唑肟钠有关物质HPLC测定方法进行优化,通过对其流动相缓冲盐种类、梯度洗脱程序、柱温等参數的调整,建立新的HPLC方法,以满足对不同企业产品质量控制的需要。
1 仪器及试药
1.1 仪器与试剂
高效液相色谱仪(岛津LC-2030 3D),紫外检测器(岛津DAD),恒温水浴锅(德国Haake),电子分析天平(瑞士Mettler Toledo官方XPE205),高纯水机(美国Millipore公司)。
乙腈(色谱纯,Sigma公司),乙酸铵(分析纯,国药试剂),高纯水(实验室自制)。
1.2 试药
头孢唑肟对照品(130504-202304),含量为97.8%,购自中国食品药品检定研究院;杂质D(QRTBZWX023-20220729),含量为98.58%,头孢唑肟开环物(QRTBZWX017-20220902)购自广州清瑞生物科技有限责任公司;头孢唑肟E异构体(20230201),来自Molcoo Chemicals Inc.;杂质3、杂质6、杂质12、杂质13、杂质16、杂质18、杂质KTO、二聚体及其异构体、开环脱羧S-氧化物1和开环脱羧-S氧化物2对照品均为西南药业股份有限公司提供;2-巯基苯并噻唑对照品(G1211642),含量为98.45%,购自Ehrenstorferf公司。
1.3 样品
76批注射用头孢唑肟钠均为2023年国家药品评价性抽验样品,涉及28个生产企业6个规格(0.25、0.5、0.75、1.0、1.5和2.0 g)的产品。头孢唑肟原料为企业提供。
2 方法
2.1 色谱条件
采用Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 μm)色谱柱;以 pH 3.6的50 mmol/L的乙酸铵溶液为流动相A,乙腈为流动相B,按表1方式进行梯度洗脱;流速为0.8 mL/min;柱温:35℃;检测波长为254 nm;进样体积:20 μL。样品临用新制。
2.2 样品制备
2.2.1 系统适用性溶液的制备
取头孢唑肟杂质18、开环脱羧S-氧化物1、开环脱羧S-氧化物2、杂质KTO、头孢唑肟开环物、杂质D、杂质3、头孢唑肟E异构体、二聚体及杂质12杂质对照品各约5 mg,分别至50 mL量瓶中,作为杂质储备液。取头孢唑肟钠原料150 mg,再分别取上述杂质储备液3 mL至100 mL量瓶中,得头孢唑肟钠1.5 mg/mL,各杂质3 μg/mL的溶液,作为系统适用性溶液。
2.2.2 对照品溶液的制备
取头孢唑肟对照品适量,精密称定,加pH7.0的磷酸缓冲液溶解并定量稀释制成每1 mL中含头孢唑肟3 μg的溶液作为对照品溶液。
2.2.3 供试品溶液的制备
临用新制。取本品适量(约含头孢唑肟75 mg),精密称定,置50 mL量瓶中,用pH7.0磷酸盐缓冲液溶解并定量制成每1 mL中约含头孢唑肟1.5 mg的溶液。
2.3 测定法
精密量取空白溶液、系统适应性溶液、对照品溶液及供试品溶液各20 ?L注入液相色谱仪,记录色谱图;采用主成分外标法以峰面积计算产品中已知杂质和未知杂质的含量。
2.4 方法验证
参考《中国药典》2020年版四部9101分析方法验证指导原则,对方法的专属性、线性和范围、准确度、检测限、定量限、精密度、耐用性以及溶液稳定性进行验证。
2.4.1 专属性
(1)未破坏溶液的制备 取本品约75 mg(以头孢唑肟计),用pH7.0缓冲液溶解并稀释至50 mL,作为供试品溶液A。
(2)强酸破坏溶液的制备 取本品约75 mg,加盐酸10滴,放置30 min,用1 mol/L氢氧化钠调节pH至中性,以 pH7.0缓冲液稀释至50 mL,作为供试品溶液B。
(3)强碱破坏溶液的制备 取本品约75 mg,加 5 mol/L氢氧化钠10滴,放置30 min,用1 mol/L 盐酸调节pH至中性,以pH7.0缓冲液稀释至50 mL,作为供试品溶液C。
(4)氧化破坏溶液的制备 取本品约75 mg,加双氧水1滴,水10 mL,放置30 min,以 pH7.0缓冲液稀释至50 mL,作为供试品溶液D。
(5)高温破坏溶液的制备 取本品约75 mg,以 pH7.0缓冲液稀释至50 mL,置100℃水浴中放置1 h,作为供试品溶液E。
(6)光破坏溶液的制备 取本品,置5000 Lx照度下放置2 d,取约75 mg,以 pH7.0缓冲液稀释至50 mL,作为供试品溶液F。
分别取上述强制降解试验溶液(A~F)20 ?L,进行专属性验证。
2.4.2 线性与范围
精密称取头孢唑肟、头孢唑肟杂质18、开环脱羧S-氧化物1、开环脱羧S-氧化物2、杂质KTO、头孢唑肟开环物、杂质D、杂质3、头孢唑肟E异构体、二聚体及杂质12对照品适量,稀释制成浓度分别为0.75、1.2、1.8、3.0、3.6和4.5 μg/mL的系列浓度溶液,分别进样;对峰面积(Y)和进样量(X)进行线性回归分析。
2.4.3 准确性
选取杂质含量已知的企业A(编号1)样品,进行加样回收率实验。首先制备每毫升分别含头孢唑肟钠杂质18(1.21 μg)、开环脱羧S-氧化物1(1.22 μg)、开环脱羧S-氧化物2(1.19 μg)、杂质KTO(1.18 μg)、杂质16(1.31 μg)、杂质6(1.27 μg)、杂质3(1.09 μg)、杂质13(1.15 μg)、二聚体(1.23 μg)、杂质12(1.03 μg)的诸杂质混合杂质储备液。精密称取约含头孢唑肟75 mg的企业A的样品9份,分别置50 mL量瓶中,再分别加入混合杂质储备液(0.9,0.9,0.9,1.5,1.5,1.5,2.25,2.25和2.25 mL),用pH7.0磷酸盐缓冲液稀释至刻度,即得。
2.4.4 LOD和LOQ
分别采用头孢唑肟、杂质C、杂质D和杂质E的线性(1)溶液(0.75 ?g/mL),逐步稀释测定,以S/N=3时的浓度作为检测限,以S/N=10时的浓度作为定量限。
2.4.5 重复性
取某厂家同一批样品,按“2.2.3”方法分别制备6份供试品溶液,测定其杂质的量,通过6份样品测定结果的差异,评价方法的重复性。
2.4.6 溶液稳定性
供试品溶液在室温放置0、1、2、4、6和8 h,分别测定头孢唑肟、最大单个杂质(头孢唑肟开环物)、二聚体及总杂质含量的变化,评价供试品溶液室温放置的稳定性。
3 结果与讨论
3.1 新HPLC方法与药典方法的比较
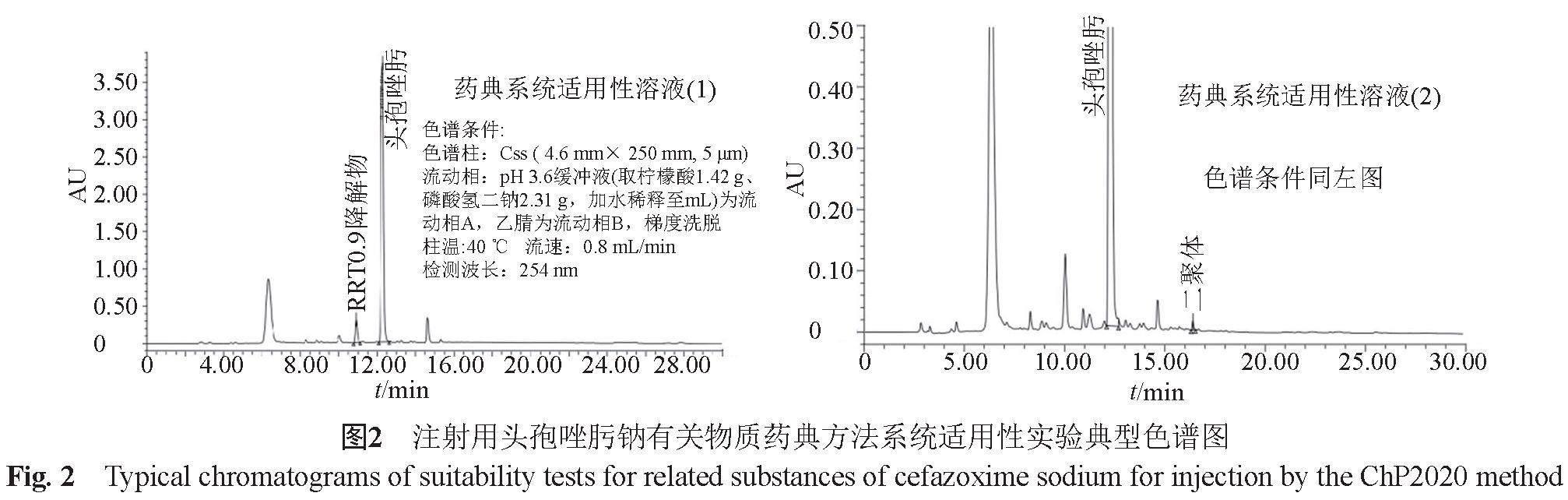
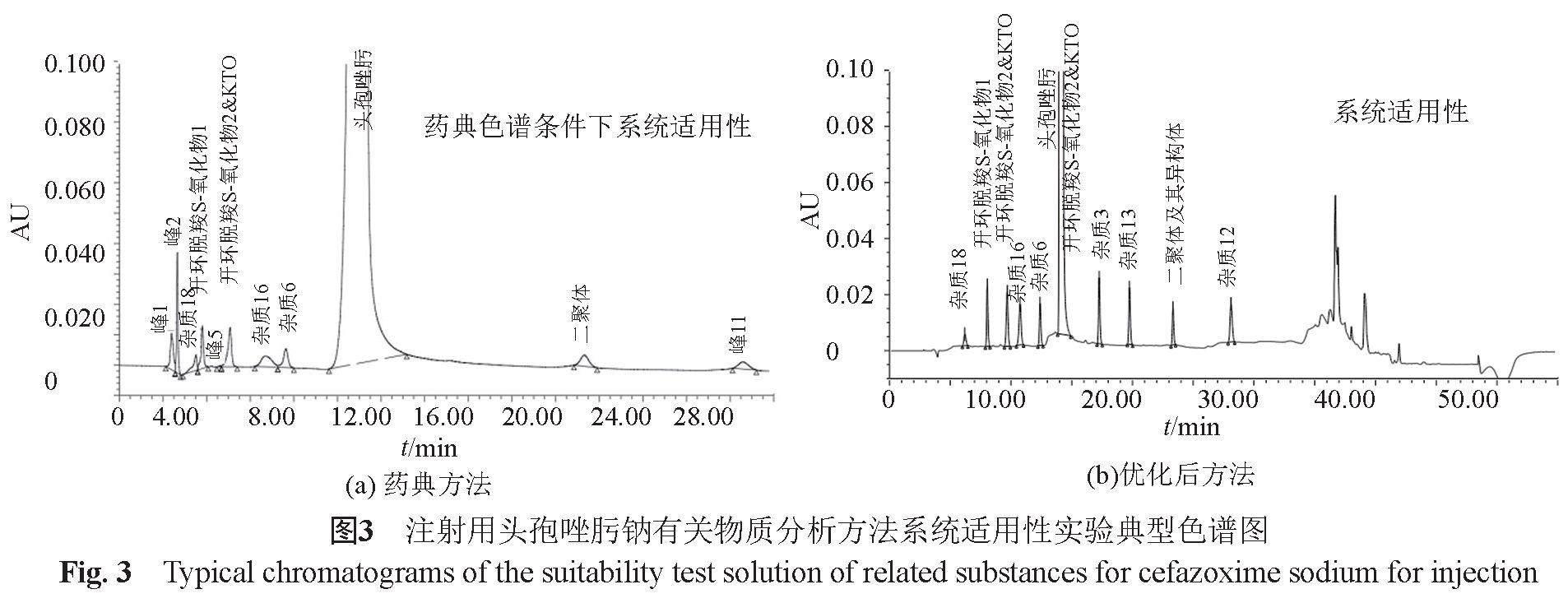
通过对检验中涉及的8个不同有关物质分析方法的比较,以诸杂质的分离情况为指针,对《中国药典》有关物质分析方法中的流动相(由柠檬酸、磷酸二氢钠缓冲盐体系改进为乙酸铵缓冲盐体系)和梯度洗脱程序(梯度时间由30 min改为55 min)和柱温(40 ℃柱温改为35 ℃)进行了优化。药典方法系统适用性典型色谱图见图2,当药典方法满足其系统适用性试验的要求时,进样优化方法的系统适用性溶液(图3a)可见,药典色谱系统仅能分离出其中的7种已知杂质(头孢唑肟杂质18、开环脱羧S-氧化物1、开环脱羧S-氧化物2和杂质KTO、杂质16、杂质6和二聚体),且杂质18与溶剂峰的倒峰分离较为困难,杂质16的峰形较差;而改进后的色谱方法对诸杂质的分离良好(图3b),且各杂质峰的峰形亦较理想。提示优化后的方法明显优于原药典方法,可满足对不同企业产品质量控制的需要。
3.2 方法学验证
3.2.1 专属性
对诸强制降解溶液进行分析(图4),强制降解试验产生的杂质种类和杂质量的变化见表2。强制降解溶液中的各杂质峰均能达到有效要求,表明新方法的专属性良好,可有效分离头孢唑肟的主成分及各种降解杂质。由表2同时可以看出与,酸破坏对样品的稳定性影响最小,仅杂质18和杂质12略有增长;碱破坏对杂质的种类和量均有较大影响,杂质16和杂质3增长最多,同时产生了新的未知杂质;高温降解主要产生杂质18和杂质3;氧化破坏主要产生杂质开环脱羧S-氧化物1和开环脱羧S-氧化物2;光破坏,杂质18和杂质6略有增加。即头孢唑肟钠溶液在碱性、高温及氧化条件下极不稳定。
3.2.2 线性和范围
头孢唑肟的线性方程为:Y=57824X+13072,r为1.000;杂质18为:Y=41930X-3664,r为0.999;开环脱羧S-氧化物1为:Y=28876X+85,r为1.000;开环脱羧S-氧化物2为:Y=34236X-1881,r为1.000;杂质KTO为Y=17118X-941,r为1.000;头孢唑肟开环物线性方程为:Y=19059X-6566,r为1.000;杂质D为:Y=39948X-1065,r为1.000;杂质3线性方程为:Y=60879X-357,r为1.000;头孢唑肟E异构体为:Y=43105X+10960,r为1.000;二聚体为:Y=23456X+7602,r为1.000;杂质12为:Y=55387X-1417,r为1.000。结果表明:头孢唑肟钠及其10种已知杂质均在0.75~4.5 μg/mL范围内線性关系良好。
3.2.3 准确性
头孢唑肟钠杂质18、开环脱羧S-氧化物1、开环脱羧S-氧化物2、杂质KTO、杂质16、杂质6、杂质3、杂质13、二聚体及杂质12的加样回收率分别为99.4%、97.4%、96.9%、99.6%、101.6%、98.6%、96.5%、97.9%和100.8%。
单个杂质在药典标准中限度要求为0.5%,二聚体为0.1%。《中国药典》2020年版四部9101药品分析方法验证指导原则[9]中规定:样品中待测成分含量为0.1%时,回收率限度要求为90%~108%。各已知杂质平均回收率范围为“96.5%~101.6%”,均符合药典要求,表明新方法的准确度良好。
3.2.4 LOD和LOQ
头孢唑肟及10种已知杂质(头孢唑肟钠杂质18、开环脱羧S-氧化物1、开环脱羧S-氧化物2、杂质KTO、杂质16、杂质6、雜质3、杂质13、二聚体及杂质12)的检出限分别为0.82、1.26、1.39、1.42、1.16、3.93、1.53、0.81、0.98、1.96及1.54 ng;定量限分别为1.63、6.30、2.78、2.84、2.31、7.86、3.06、1.62、1.90、3.87及3.09 ng。
3.2.5 重复性
取某厂家同一批号6份样品进行测定,杂质测定结果分别为头孢唑肟钠杂质18未检出、开环脱羧S-氧化物1未检出、开环脱羧S-氧化物2及杂质KTO未检出、杂质16检出0.19%(RSD6.6%)、杂质6检出0.14%(RSD10.4%)、杂质3未检出、杂质13未检出、二聚体及其异构体检出0.11%(RSD6.8%)杂质12未检出,其他未知最大杂质检出0.18%(RSD8.6%)、总杂质检出0.22%(RSD8.4%),RSD均符合要求,表明新方法重复性良好。
3.2.6 溶液稳定性
供试品溶液在室温随着放置时间的延长,头孢唑肟含量变化不显著,RSD为0.28%;二聚体稍有增多;头孢唑肟开环物及总杂质明显增大,6 h后超出限度范围,头孢唑肟开环物由最初的0.14%增长至0.48%,总杂质由最初的0.15%增长至0.49%,RSD分别为48.8%和46.8%。即注射用头孢唑肟钠溶液室温下稳定性较差,建议临用新制。
3.2.7 耐用性
采用C18色谱柱(Waters Symmetry Shield RP18 250 mm×4.6 mm, 5 μm)替代Agilent ZORBAX SB-C18,同时改变流动相的流速(0.7和0.9 mL/min)和柱温(30和40 ℃),以及流动相中色谱纯乙腈的品牌头孢唑肟钠及各已知、未知杂质均可有效分离。
3.3 样品测定
通过对28家企业76批样品的测定结果进行分析,典型色谱图见图5,发现各生产企业样品检出的杂质种类和数量均比药典多和高。
28家生产企业中,参比企业“西南药业”样品中杂质16的检出量约为0.17%;“山东X企业、东北X企业及珠海X企业”3个企业检出的杂质16相对较大,范围约在0.33%~0.43%之间;“重庆、海口、上海、苏州、河北及汕头等”7个企业杂质16检出较小,范围约在0.15%~0.20%之间,基本与参比制剂一致;其余17个生产企业杂质16的检出范围约在0.20%~0.30%之间。
28家生产企业中,参比企业“西南药业”样品中总杂质的检出量约为0.23%;“山东X企业、东北X企业及珠海X企业”3个企业检出的总杂质相对较大,范围约在0.43%~0.58%之间;“重庆、海口、上海、苏州、河北及汕头等”7个企业总杂质检出较小,范围约在0.20%~0.26%之间,基本与参比制剂一致;其余17个生产企业总杂质检出范围约在0.30%~0.40%之间。
二聚体及其异构体的检出量主要分布区域为0.005%~0.018%的区域,远小于限度0.10%,且各企业之间检出量无明显差异,基本与参比企业一致。
4 结论
本文所建立的注射用头孢唑肟钠有关物质方法可满足对不同企业产品质量控制的需要。在注射用头孢唑肟钠中可检出10余种杂质,但主要杂质(超过0.1%)为杂质16(头孢唑肟开环物),将其作为特定杂质控制具有重要意义;同时,二聚体体现了产品中聚合物的水平,具有重要的控制意义。根据对实际样品的考察结果,杂质16、二聚体及总杂质分别分布在0.2%,0.02%和0.3%左右,可据此制定杂质的限度。
参 考 文 献
郑洁, 隆莉, 袁帅. 临床常用抗菌药治疗肺炎克雷伯菌感染的临床疗效[J]. 国外医药(抗生素分册), 2021, 42(5): 286-289.
施毅, 苏欣, 肖永营, 等. 注射用头孢唑肟钠治疗细菌性感染的多中心随机对照临床研究[J]. 中国抗感染化疗杂志, 2005, 5(6): 350-354.
戎霞君, 陈巍, 黄绍光. 注射用头孢唑肟钠治疗下呼吸道细菌感染的研究[J]. 世界临床药物, 2006, 27(7): 395-400.
黄美英, 夏星, 罗凯, 等. 头孢唑肟钠生产工艺及条件的研究[J]. 中国药房, 2007, 18(22): 1695-1697.
国家药典委员会. 中华人民共和国药典[S]. (2020年版二部). 中国医药科技出版社, 2020: 342-344.
杨倩, 李伟, 曹晓云, 等. 注射用头孢唑肟钠的杂质谱研究[J]. 中国药学杂志, 2014, 49(19): 1750-1754.
孟君, 林顺权, 蒲含林, 等. 头孢唑肟钠有关物质的研究(英文)[J]. 中国抗生素杂志, 2022, 47(7): 682-690.
于佳, 张一竹, 李委佳, 等. 头孢唑肟钠混合降解杂质对照物的研究与应用[J]. 化学工程师, 2018, 32(2): 8-10.
国家药典委员会. 中华人民共和国药典[S]. (2020年版四部). 中国医药科技出版社, 2020: 480-483.
