
蛋白质二级结构在线服务器预测评估
2019-04-24 06:12朱树平刘毅慧
生物信息学 2019年1期
朱树平,刘毅慧
(齐鲁工业大学(山东省科学院) 计算机科学与技术学院,济南 250353)
蛋白质是人体的有机大分子,是生命活动的主要承担者,在生物信息学领域,一直致力于对于蛋白质的研究。为了研究蛋白质的功能,往往从结构入手,但蛋白质结构有多种,其中关于二级结构的研究,有助于发现三维立体结构和提供蛋白质功能注解,因此大多数人都致力于蛋白质二级结构的研究。
在1951年,鲍林和科里首次提出了关于蛋白质二级结构问题[1],最初对于蛋白质二级结构的预测方法主要是通过研究氨基酸序列来进行,准确率在60%左右。Rost[2-3]等人在研究中采用PHD算法,把多序列排列中包含的进化信息作为神经网络的输入,预测蛋白质的二级结构准确率超过了70%。Zafer[4]等人使用动态贝叶斯分类器的稀疏算法,得到了76.3%的准确率。Kurniawan[5]等人使用SVM结合位置特异性打分矩阵(Position-specific scoring matrices,PSSM)和蛋白质结构的物理化学特征来预测,准确率达到80%左右。Wang[6]等人通过结合PSSM和氨基酸序列信息,并使用一种称为二级结构递归编码器-解码器网络(SSREDN)来解决输入蛋白质特征与SS之间的序列-结构映射关系,使用CullPDB和CB513数据库测试,分别达到84.2%,82.9%的Q3准确率。蛋白质二级结构预测方式不断注入新的活力,现在很多方法都实现了在线服务器的预测,本文选取了PSRSM、MUFOLD、SPIDER、RAPTORX、JPRED和PSIPRED 6种服务器,分别阐述其算法原理,并通过测试数据比较每一个的预测准确度,从而给出当前在线服务器二级结构的评估。
1 在线服务器原理
1.1 PSRSM
该服务器使用基于数据分区和半随机子空间(Partition and semi-random subspace method,PSRSM)的方法[7]。在传统的随机子空间方法中,低维子空间是由高维空间随机采样产生的,PSRSM使用的半随机子空间方法能够有效的保证基础分类器的准确性和多样化。该方法的主要步骤如下:首先把训练数据根据蛋白质的长度划分为不同的子集合,建立模型;然后使用半随机子空间的方法生成子空间,并在子空间上训练基础分类器;最后根据多数投票的规则,在子集上把分类器结合起来,生成最终的分类器,其中使用SVM作为最基本的分类器。
具体来说,对于输入使用PSI-BLAST程序生成PSSM数据,并且PSI-BLAST使用BLOSUM62进化矩阵搜索NCBI的非冗余(NR)数据库的缩减版本,按照上述原则得到的PSSM是20*L的矩阵,20为氨基酸的个数,L为每个蛋白质的长度。在实验中使用13个滑动窗口来获取蛋白质序列信息和预测序列中心的蛋白质二级结构。假设输入一个长度为L的蛋白质,会产生260*L(13*20*L)的输入矩阵。从260个特征值选取160个作为主要特征,作为网络输入。最后建立12个分类器进行训练。那么一个新的蛋白质序列会根据其长度,选择合适的分类器进行预测。
实验的训练集选取了ASTRAL数据集的6 892条蛋白质数据和CullPDB数据集的12 288条蛋白质数据,去掉相似度较高的蛋白质后,训练集总共包括15 696条数据。测试集使用99个CASP10数据、81个CASP11数据、19个CASP12数据、513个CB513数据、1 673个25PDB的数据和2018年2月1号之前的100条数据(T100),实验得到使用6个GTPCs模型在25PDB、CB513、CASP10、CASP11、CASP12和T100数据中的蛋白质二级结构的Q3预测准确率分别是86.38%、84.53%、85.51%、85.89%、85.55%和85.09%。该服务器预测蛋白质序列范围是10到800,预测网址为:http://qilubio.qlu.edu.cn:82/protein_PSRSM/default.aspx。
1.2 MUFOLD
MUFOLD采用的是一种名为深度初始-内部-初始(Deep 3I)的新型网络来预测蛋白质二级结构,并且对于输入的特征矩阵做了细致考量,特征矩阵中结合了氨基酸的理化性质、PSI-Blast特征和HHBlits特征[8]。其中对于理化性质的特征矩阵,设置了从-1到1之间选取的8个数字来表示一个氨基酸,前7位表示氨基酸理化性质,后一位用1或0表示是否输入氨基酸。如表1,“*”表示某一类氨基酸,“n”表示依据理化性质设置的数值。MUFOLD设置默认输入矩阵为700*8,若假设输入一个氨基酸序列个数为600的蛋白质,设置矩阵时会把前600行的前7位按照本身理化性质设置,第8位设为0,而后100行的前7为全部设为0,后一位设置为1。
对于PSI-Blast的特征,按照类似原理用从0到1的选取21位数字表示一个氨基酸,前20位根据得到的PSSM值设置,后一位用1或0表示是否有输入;对于HHBlits特征则用0到1之间的31位数字表示一个氨基酸,前30位根据HMM文件设置,最后一位同样用0或1表示输入。以上三个特征被组合成一个58位的特征,作为网络的输入。
Deep3I网络是由2个Deep3I块、一系列卷积和完全联通的致密层构成。而Deep3I块是由初始模块递归嵌套构成,初始模块通过卷积操作能够有效提取氨基酸残基之间的非局部相互作用。Deep3I网络通过用TensorFlow和Keras不断进行训练和实验来对蛋白质二级结构进行预测。
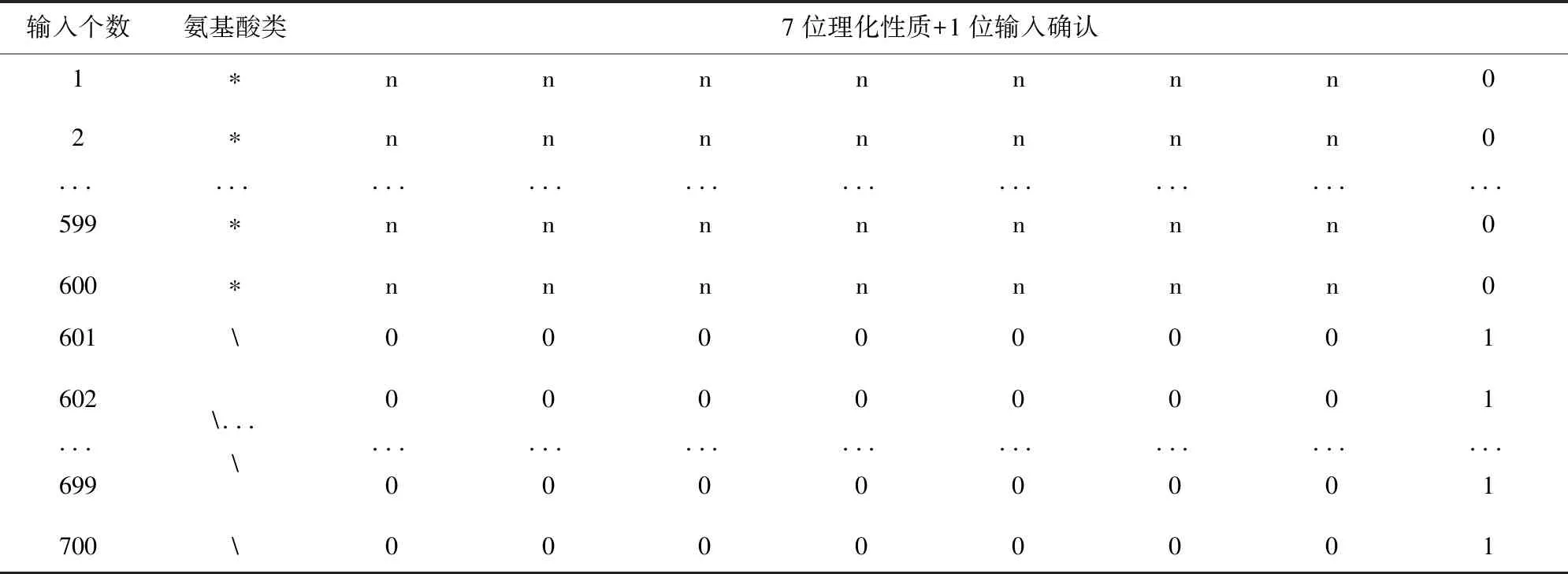
表1 按照氨基酸理化性质设置的输入矩阵Table 1 Input matrix set according to the physical and chemical properties of amino acids
MUFOLD实验中的数据集使用蛋白质序列长度介于50到700之间的数据,来自CullPDB、JPRED、CASP、CB513和PDB 5个公开的蛋白质数据库。具体来说:从CullPDB选取了9 581条数据,其中随机选出9 000条作为训练集,剩下的581条作为测试;从JPRED选取的数据均来自不同的超级家族;CASP的数据集经过筛选后CASP10的98条数据,CASP11的83条数据,CASP12的40条数据被使用;CB513和385条PDB数据也同样被应用于MUFOLD的实验中。MUFOLD测试数据的范围是30到700,测试网址是:http://mufold.org/mufold-ss-angle/。
1.3 SPIDER
Hefferman[9]等人提到对于蛋白质二级结构预测和溶剂接触表面积的研究,多年一直停滞不前的原因来自于,有些氨基酸残基在三维结构中距离很近而在蛋白质序列中距离很远,因此较难捕获氨基酸残基之间的非局部相互作用。现有的机器学习的方法基本都使用10~20个滑动窗口来获取氨基酸的相互作用。而SPIDER不使用滑动窗口,采用一种长期短期记忆(Long Short-Term Memory , LSTM)双向递归神经网络(Bidirectional Recurrent Neural Networks ,BRNNs)的机器学习模型来实现预测,能够捕捉氨基酸残基之间的非局部相互相互作用,实验证明它能够改善蛋白质二级结构、骨干角度、接触号码和溶剂可及性的预测。
该网络的LSTM-BRNN模型是由两个使用LSTM细胞的BRNN层和两个紧密连接用整流线性单元(Rectified Linear Unit, ReLU)激活的隐含层构成,它被用于四次迭代中。对于该网络的输入,包含了7种具有代表性的蛋白质氨基酸理化性质(Physio-chemical properties,PP)、20维来自PSI-Blast的PSSM和30维来自HHBlits每个残基的隐藏马尔科夫模型的序列谱(HMM Profiles),把这些数据放入由LSTM-BRNNs网络构成的迭代中,进行四次迭代(其中一次迭代包括两个LSTM-BRNN),最后得到最终机器学习模型。该过程主要结构如图1所示。在训练期间为防止过拟合,使用丢失率为50%的丢失算法,并用Adam优化训练过程,该网络能够在不使用滑动窗口的条件下捕获长短距离交互。
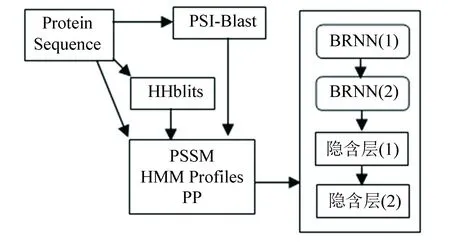
图1 SPIDER 主要结构Fig.1 Main structure of SPIDER
1.4 RAPTORX
RAPTORX使用由深度卷积神经网络(Deep convolutional neural network , DCNN)和条件随机场(Conditional random fields,CRF)组合而成的深度卷积神经场(Deep Convolutional Neural Fields,DCNF),来预测蛋白质二级结构,并且对网络采用一种在ROC曲线下面积的(Area under the ROC curve,AUC)最大化方法来训练,从而能够很好地解决紊乱序列蛋白质的预测问题[11]。Wang[12]提到在使用蛋白质序列文件后,RAPTORX在数据集CASP和CAMEO能够得到大约为84%的Q3准确率和72%的Q8准确率,不使用序列文件能够获得约为74%的Q3准确率和59%的Q8准确率,它能够有效的解决复杂的基因结构关系建模和相邻残基间的建模。Wang[13]指出DCNF使用DCNN代替CNF中使用的浅层神经网络,能够捕获输入和输出标签之间复杂的关系,并且能够捕获远程的序列信息。
RAPTORX实验中使用的数据有6 125个CullPDB数据,CB513数据、123个CASP10数据、105个CASP11数据和CAMEO的数据,还有JPRED公开的1 338个训练数据和149个测试数据。RAPTORX测试数据范围是26到4 000个蛋白质序列,预测网址为:http://raptorx.uchicago.edu/StructurePropertyPred/predict/。
1.5 JPRED
JPRED服务器从1998年开始提供蛋白质的预测到现在已经发展到JPRED4版本。JPRED3版本用JNET算法提供单个蛋白质序列或者多序列比对(MSA)的预测,其中JNET使用JNET v2.0。JNET v2.0不使用频率文件,只使用PSI-BLAST的PSSM配置文件和HMMER的隐马尔可夫模型,把神经网络由9个单元增加到100个单元,该方法是通过对超家族级别的SCOPe数据的Astral汇编衍生的序列和结构非冗余数据集进行7倍交叉验证培训而开发的[14],最后使用149条盲数据进行测试得到了81.5%的Q3准确率。
JPRED4版本和JPRED3一样,同样使用JNET算法并提供单一序列和多序列比对的蛋白质序列的二级预测。不同的是它选取1 358个SCOPe/ASTRAL v.2.04 超级家族中的一个为代表,用JNET 2.3.1进行7倍交叉验证的实验,通过寻找UniRef90 v.2014_07来生成PSI-BLAST文件并为每一个蛋白质序列建立多重序列比对。最后在150个训练集上获得了82%的准确率[15]。同时JPRED在线服务器也可以提供溶剂可及性和卷曲螺旋区的预测,预测网址为:http://www.compbio.dundee.ac.uk/jpred4/index.html。
1.6 PSIPRED
Mcguffin[16]等人指出PSIPRED服务器结合了三种先进的技术,分别是PSIPRED、GenTHREADER和MEMSAT 2。其中PSIPRED采用严格的交叉验证程评估性能,并且采用两个前馈的神经网络,对从PSI-BLAST获得的输出进行分析,从而得到可靠的二级结构预测结果;GenTHREADER用来推断跨膜蛋白的结构和拓扑结构;MEMSAT2能够快速识别蛋白质的折叠信息,预测网址为:http://bioinf.cs.ucl.ac.uk/psipred/。
从以上6个服务器预测过程的角度分析,可以看到每个服务器各有优缺点。其中能够批量上传和下载实验结果的是PSRSM、SPIDER3和RAPTORX,给定结果为压缩包的形式,需进一步整合。服务器JPRED和PSIPRED都必须遵循每次只能上传一个蛋白质文件(或序列)的约定,而且结果是以邮件的形式发送到邮箱里面,并且PSIPRED在同一时间段内最多只允许上传20条数据进行预测,因此预测结果获取过程较为复杂。MUFOLD虽然网站上说明一次可以批量上传少于10条的数据但是在实验中获取数据,最多一次只可上传4条数据进行预测。6个服务器预测的时间相差并不是很大,主要在于预测结果的获取方式上存在很大差距。
2 数据选取和评估标准
基于每个服务器都可以预测为前提,依据蛋白质发布的月份和其同源性分别选取了150条数据进行实验,并采用了合适的评价标准来评估。
2.1 数据选取
数据选取遵循以下原则:数据选取2018年PDB最新发布的数据,保证了测试集不在服务器的训练集中;数据来自不同的时间段,更具有分散性;数据量较大,使得实验结果更具有说服力;选取的蛋白质长度能够让每一个服务器都可以进行测试,并得到预测结果。基于上述的条件从2018年4、5、6月份分别选取了50条蛋白质序列进行第一次实验,数据选取如表2所示。
并且为了使实验结果更具有可靠性,又进一步从2018年4到8月,基于同源性的30%,50%和70%随机分别选取了50条数据,共150条数据(T150)进行第二次实验,该实验的数据选取如表3所示。
2.2 评估标准
本文采用了两种衡量蛋白质二级结构预测准确性方法:Q3和Sov的值主要是衡量个别残基分配的精度,Sov的值主要是衡量全元素的预测精度。
2.2.1Q3
按照DSSP[17]的规定,通常我们把蛋白质二级结构划分为H、G、I、E、B、T、S和-,8种状态。而这8这种状态,按照H、G、I→H,E、B→E,其他→C的方式,将一条氨基酸序列转化为H(螺旋)、E(折叠)、C(卷曲),3种状态。则Q3表示被正确预测的三种状态的氨基酸数占整个氨基酸序列的比例。符合以下计算公式:
(1)
其中:SE是E类蛋白质结构准确预测的数量,SH是H类蛋白质结构准确预测的数量,SC是C类蛋白质结构准确预测的数量,S是指总的氨基酸数量,Q3指的是三种状态下,蛋白质二级结构预测的准确率。

表2 DB150 数据集Table 2 DB150 data set

表3 T150 数据集Table 3 T150 data set
2.2.2Sov
Sov的计算是基于重叠片段比值的一种测度,它对预测结果和观察到的结果同等对待。同样按照上述Q3的思想把蛋白质二级结构划分为螺旋、折叠和卷曲三种状态。如果假设观察到的序列记为S1,预测到的序列记为S2,S0为S1和S2所有状态相同的片段,那么S0必定会包含一对重叠和一个螺旋,接下来S1的长度为length(S1),并且把每对中S1和S2序列个数求并集记为max(S1,S2),把S1和S2的序列个数求交集记为min(S1,S2)。在上述基础上把Sov的计算公式定义为[18]:
(2)
其中关于δ的设定是为了允许蛋白质结构中边缘处片段的变化,δ(S1,S2)取值符合以下定义:
(3)
3 实验及结果
从PDB中下载得到最新的蛋白质数据,然后分别上传到6个预测服务器上进行测试。上传蛋白质序列得到的预测结果后,通过与正确的三态的DSSP结果相比较,计算每一条蛋白质的Q3和Sov准确率。第一次实验中每月数据和DB150的Q3和Sov准确率如表4所示。第二次实验中基于30%,50%,70%的同源度数据和T150的Q3和Sov的实验结果如表5所示。
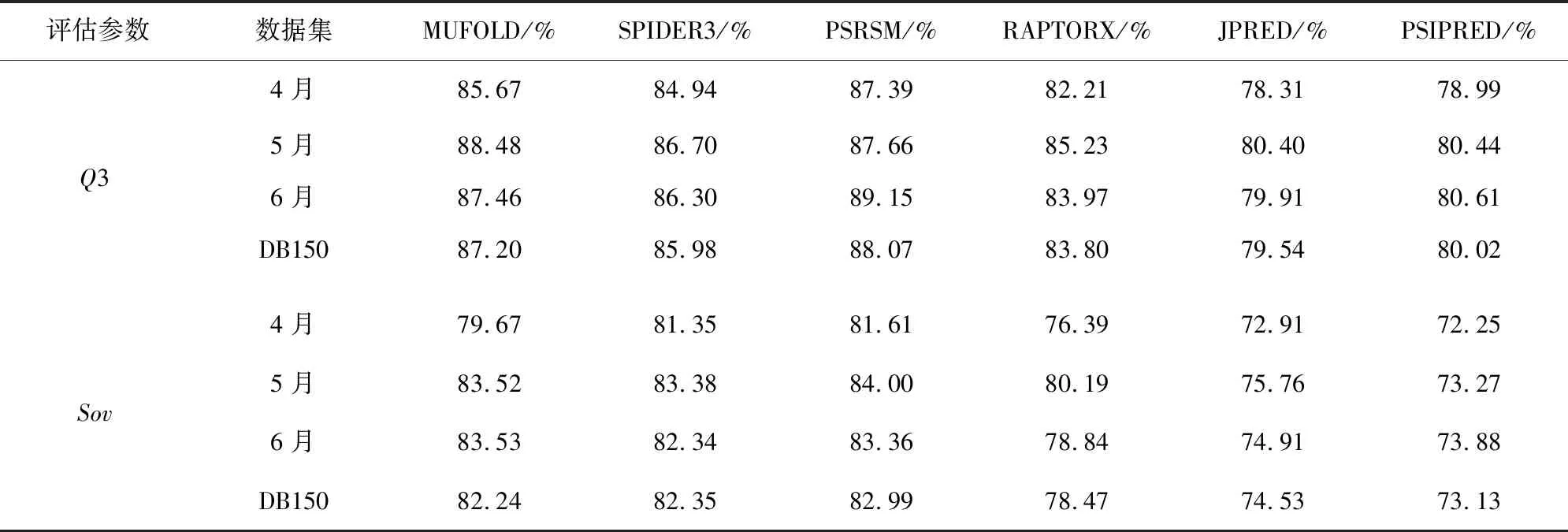
表4 实验1的Q3和 Sov平均准确率Table 4 Average accuracy of Q3 and Sov in Experiment 1
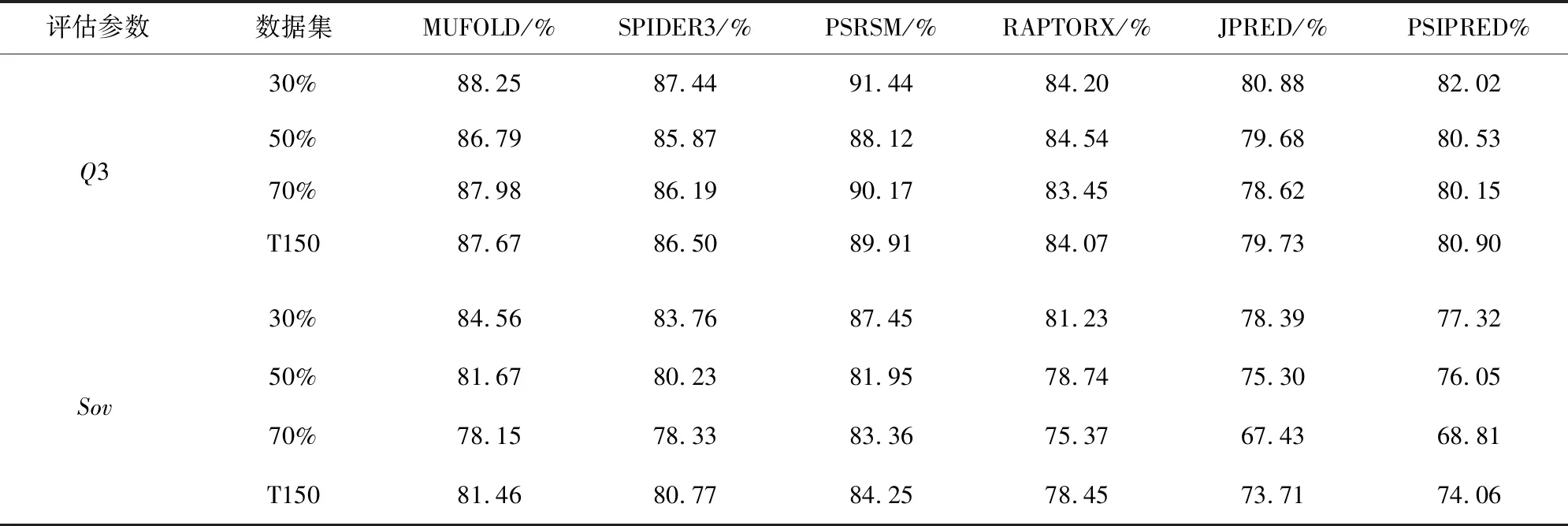
表5 实验2的Q3和Sov平均准确率Table 5 Average accuracy of Q3 and Sov in Experiment 2
从实验结果中看到,不论是基于月份的蛋白质数据,还是基于同源性不同划分的数据,PSRSM都取得了在同一类别中较好的效果,Q3的预测准确率有时甚至超过90%。按照月份划分时,4月份的数据集中,PSRSM达到了最好的预测效果,Q3和Sov的值分别为87.39%和81.61%;在5月份数据集中,MUFOLD的Q3准确率最高,为88.48%,Sov准确率仅次于PSRSM的84.00%,为83.52%;在6月份数据集中PSRSM的Q3获得最高准确率为89.15%,而Sov仅次于MUFOLD的83.53%,为83.36%。在综合数据DB150的结果中我们得到6种预测方式Q3的准确率由高到低为PSRSM的88.07%,MUFOLD的87.20%,SPIDER的85.98%,RAPTORX的83.80%,PSIPRED的80.02%和JPRED的79.54%;Sov准确率由高到低为PSRSM的82.99%,SPIDER3的82.35%,RAPTORX的78.47%,JPRED的74.53%和PSIPRED的73.13%,PSRSM得到了Q3和Sov的最高准确率。
在基于同源性的实验中,结果显示基于30%时,PSRSM得到了91.44%的Q3准确度和87.45%的Sov准确度,比其他服务器中最好的MUFOLD分别高出3.19和2.89个百分分点;同源度为50%时,PSRSM的Q3为88.12%,Sov为81.95%,分别比MUFOLD高出1.33和0.28个百分点;70%的同源度时PSRSM的Q3和Sov分别为90.17%和83.36,Q3比其他服务器中最好的MUFOLD高出2.19%,Sov比预测结果最好的SPIDER高出5%。总体来看在T150中Q3和Sov准确率由高到低分别为PSRSM的89.91和84.25%,MUFOLD的87.67%和81.46%,SPIDER的86.50%和80.77%,Raptorx的84.07%和78.45%,PSIPRED的80.06%和74.06%,JPRED的79.73和73.71%。
无论在哪一种情况下,PSRSM、MUFOLD和SPIDER3都得到了超过84.9%的Q3准确率和超过78.1%的Sov准确率,其中PSRSM表现出良好的预测性能。
4 结 论
蛋白质二级结构预测的准确度,将决定人类对于蛋白质功能的了解程度。本文介绍了现在6个热门的预测服务器原理,并使用最新的数据对其二级结构预测的准确率进行评估。比较6个服务器的预测方法和实验结果,可以看到它们的研究方法都在着重解决那些三维结构中距离近而序列中距离远的氨基酸残基的预测问题,并为此一再提出新的解决思路。
PSRSM在上述实验数据中大多都取得了最好的实验结果,特别是在基于同源性差异的实验中,当同源度较很低为30%时,其Q3准确率比其他服务器中最好的MUFOLD高出3.19%,这更说明PSRSM具有更好的预测效果。PSRSM与其他服务器比较,其优点在于基于蛋白质长度划分设计模板的使用,另一点在于训练数据量非常庞大,当然也采用了合理的预测方法。通过该实验和结果也可以看出,其他服务器能否获得优越的结果与其训练数据量的大小密切相关,当然还与其各自使用的深度学习算法有关。因此今后对于蛋白质二级结构预测的研究应当重点从大数据、模板和深度学习的角度进行突破。
猜你喜欢
健康之家(2021年19期)2021-05-23
医学食疗与健康(2021年27期)2021-05-13
农业科技与信息(2021年2期)2021-03-27
中国洗涤用品工业(2019年4期)2019-05-11
中国交通信息化(2018年5期)2018-08-21
中成药(2018年1期)2018-02-02
电子技术与软件工程(2017年14期)2017-09-08
计算机应用(2017年4期)2017-06-27
中成药(2017年3期)2017-05-17
航天返回与遥感(2014年5期)2014-07-31
