
三苯胺基D-(D´-A)n型醛类化合物的合成、表征及光学性质研究
2019-12-04 11:33骆启培杨周华田志美徐华杰刘昭第
阜阳师范大学学报(自然科学版) 2019年4期
骆启培,刘 悦,杨周华,田志美,徐华杰,刘昭第
(阜阳师范大学 化学与材料工程学院,安徽 阜阳 236037)
三苯胺及其衍生物具有独特的“螺旋桨式”构型及优异的供电子和电子转移能力,其常作为较强的Donor(D)基团和富电子中心被广泛应用于制备光电材料[1-5]。而三苯胺基醛类化合物是一类重要的有机发光材料中间体[6-7],可通过Wittig反应、Wittig-Horner反应、醇醛缩合反应如Claisen-Schmidt反应、Knoevenagel反应、Perkin反应引入双键,进而增加共轭体系的长度,可有效的增加其光学性质;同时以三苯胺为核易于构建一枝、二枝、三枝化合物,多枝化效应也是增加发光性质的有效方法[8-12]。本文以卤代三苯胺、5-醛基-2-噻吩硼酸为原料,[1,1-双(二苯基磷)二茂铁]二氯化钯为催化剂,通过Suzuki偶联反应,制备了D-(D'-A)n型醛基化合物L1、L2、L3见图1,其中D为富电子三苯胺、D'为协同给电子噻吩、A为吸电子(Accept)醛基、n为 1~3。它们的结构经1H NMR,13C NMR,MALDI-TOF MS,IR 表征,并利用紫外-可见分光光度仪、荧光光谱仪,初步研究了其光学性质及构效关系。
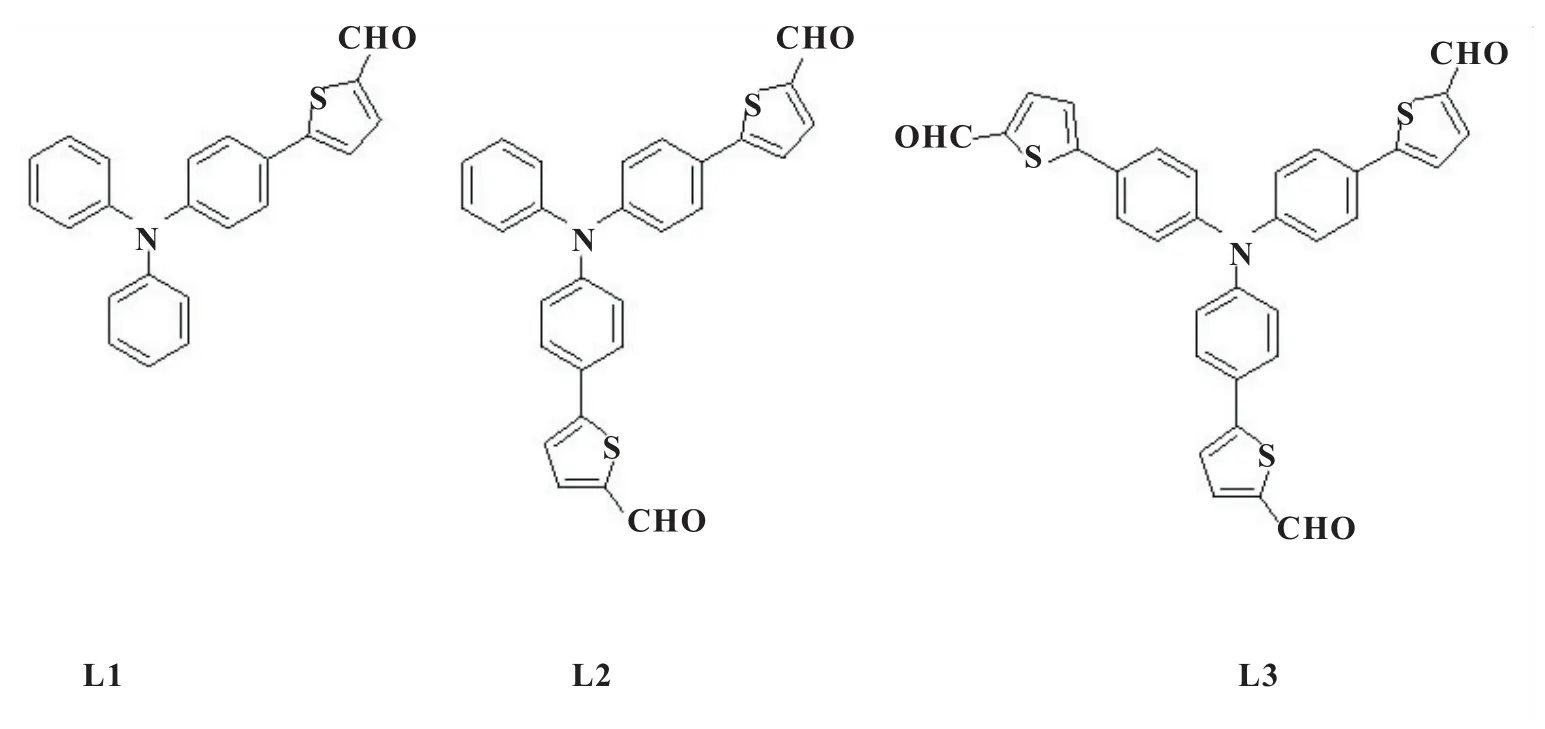
图1 D-(D'-A)n型醛基化合物L1、L2、L3的结构
1 实验部分
1.1 仪器与药品
紫外-可见分光光度计(UV-8000A)、荧光光谱仪CARY-Eclipse、Nicolet FT-IR-870SX红外分析仪(KBr压片)、Bruker 400 Ultrashield核磁共振仪,以TMS为内标、AXIMA-CFR plus型飞行时间质谱仪。
4-溴三苯胺、4,4'-二溴三苯胺、4,4',4''-三碘三苯胺、碳酸钾,PdCl2(dppf)([1,1-双(二苯基磷)二茂铁]二氯化钯)、5-醛基-2-噻吩硼酸,反应用试剂均为分析纯,光谱测试用试剂均为色谱纯。
1.2 L1、L2和L3的合成
1.2.1 L1的合成
在100 mL三口烧瓶中,依次加入4.073 g(30 mmol)碳酸钾,1.44 g(4.5 mmol)中间体 0.468 g(3 mmol)5-醛基-2-噻吩硼酸,加入 60 mL 甲苯/甲醇(v:v=1:1)搅拌使其溶解。在氮气氛围下,加入0.348 g PdCl2(dppf)(0.3 mmol),加热,避光,回流反应22 h。减压蒸出溶剂,用二氯甲烷和水萃取3~5次,合并有机相,加入约2 g无水硫酸镁干燥,静置2~3 h。抽滤,收集滤液,浓缩、拌样。柱层析后,产物用无水乙醇重结晶,得黄色固体,干燥后称重,最终获得纯产物0.91 g产率为85.1%。1H NMR(400 MHz,Acetone)δ:9.91(s,1H),7.94(d,J=4.0 Hz,1H),7.69(d,J=8.7 Hz,2H),7.56(d,J=3.9 Hz,1H),7.37(t,J=7.9 Hz,4H),7.20-7.10(m,6H),7.05(d,J=8.7 Hz,2H);13C NMR(100 MHz,DMSO) δ:184.21,153.38,149.02,146.88,141.33,140.00,130.26,127.90,125.98,125.46,124.58,124.46,122.15;MALDI-TOF MS(M+1=356.11);IR(KBr,cm-1)selected bands:2 359(w),1 662(s),1 591(s),1 489(s),1 448(s),1 329(m),1 284(m),1 228(m),1 057(m),808(m),752(m),698(s),492(w)[13]。
1.2.2 L2的合成
在250 mL三口烧瓶中,依次加入15 g(108 mmol)碳酸钾,2.5 g(6.2 mmol)4,4'-二溴三苯胺、4.4 g(28 mmol)5-醛基-2-噻吩硼酸,加入 60 mL 甲苯/甲醇(v:v=1:1)搅拌使其溶解。在氮气氛围下,加入 1.5 g PdCl2(dppf)(1.29 mmol),加热至 95 ℃,避光,回流反应22 h左右。减压蒸出溶剂,用二氯甲烷和水萃取3~5次,合并有机相,加入约2 g无水硫酸镁干燥,静置2~3 h。抽滤,收集滤液,浓缩、拌样。柱层析后,产物用无水乙醇重结晶,得黄色固体,干燥后称重,最终获得纯产物1.62 g产率为 60.9%。1H NMR(400 MHz,CDCl3)δ:9.87(s,2H),7.72(d,J=3.9 Hz,2H),7.57(d,J=8.4 Hz,4H),7.34(t,J=7.2 Hz,2H),7.15(dd,J=14.1,8.4 Hz,4H);13C NMR(100 MHz,CDCl3)δ:182.55,154.05,148.28,146.52,141.79,137.51,129.75,127.46,125.79,124.75,123.75,123.25;MALDI-TOF MS(M+1=466.09);IR(KBr,cm-1)selected bands:2 924(w),2 027(w),1 672(s),1 657(s),1 593(m),1 491(w),1 46(s),1 333(w),1 288(m),1 234(m),1 117(w),1 057(w),918(m),823(m),798(m),488(w)。
1.2.3 L3的合成
在250 mL三口烧瓶中依次加入0.5 g(3.6 mmol)碳酸钾,0.27 g(0.435 mmol)4,4',4''-三碘三苯胺,2.3 g 5-醛基-2-噻吩硼酸,在氮气氛围下,称取1 g PdCl2(dppf)加入到三口瓶中,避光,回流反应22 h。减压蒸出溶剂,用二氯甲烷和水萃取3~5次,合并有机相,加入约2 g无水硫酸镁干燥,静置2~3 h。抽滤,收集滤液,浓缩、拌样。柱层析后,产物用无水乙醇重结晶,得黄色固体,干燥后称重,最终获得纯产物1.88 g,产率为56.4%。1H NMR(400 MHz,DMSO)δ:9.90(s,3H),8.04(d,J=4.0 Hz,3H),7.87-7.75(m,6H),7.70(d,J=4.0 Hz,3H),7.24-7.11(m,6H);13C NMR(100 MHz,DMSO)δ:183.81,152.27,147.03,141.44,139.33,127.71,124.68,124.47;MALDI-TOF MS(M+1=576.07);IR(KBr,cm-1)selected bands:2 029(w),1 660(s),1 591(m),1 442(s),1 323(m),1 227(m),1 136(m),1 057(w),903(w),804(w)[14]。
2 结果与讨论
2.1 合成
由 L1~L3的1H NMR 和13H NMR 如图 2~4。可知,1H NMR的化学位移值均大于7,这是由于分子中的氢在苯环、噻吩环和醛上,其中醛上的氢处在羰基平面的去屏蔽区,共振信号向低场区移动。因此,单峰δ:9.91,9.87,9.90分别为L1-L3醛基氢的化学位移。同时具有D-(D´-A)n型的分子,当n=3时,分子L3具有较高的对称性,无论在1H NMR还是13H NMR上,产生较少的峰。
图5为 L1~L3 的红外光谱图,1 662,1 672,1 660 cm-1处的峰分别是L1~L3的醛基中的C=O伸缩振动产生的峰,1 591,1 593,1 591 cm-1处的峰分别是L1~L3碳碳双键伸缩振动产生的峰,1 228,1 234,1 227 cm-1处的峰分别是 L1~L3 的C-S伸缩振动产生的峰。综上,D-(D´-A)n型分子,官能团种类相同,只是含有的取代基数目不一样,所以三个物质的红外光谱很相似。

图2 L1在d6-CH3COCH3的1H NMR(上)和13H NMR(下)

图3 L2在d3-CH3Cl的1H NMR(上)和13H NMR(下)

图4 L3在 d6-CH3COCH3的1H NMR(上)和13H NMR(下)
2.2 光学性质
在不同溶剂中的紫外-可见分光光谱及荧光光谱测试的样品浓度均为1.0×10-5mol/L,荧光光谱测试的L1~L3的激发波长分别是425 nm,430 nm,436 nm,狭缝宽度2.5 nm。
由图6可知,L1、L2、L3分别在极性不同的溶剂中的吸收光谱相似,且它们都有两个吸收峰(L1:297 nm,398 nm;L2:290 nm,417 nm;L3:293 nm,414 nm),其中短波290 nm处的吸收峰是三苯胺基团的特征吸收峰,长波大约400 nm处的吸收峰为分子内Donor基团向Acceptor基团电荷转移引起的吸收峰。由图7可以看出L1、L2、L3在THF中,短波处约290 nm的吸光度差别不大,长波处约400 nm的最大吸收随醛基的增多,最大吸收波长逐渐红移,吸光度随n的增加(n=1时,A=0.404;n=2 时,A=0.630;n=3 时,A=0.831),吸光度逐渐增加;在相同的浓度下,n每增加1,吸光度增加约0.2,这是由于随着枝数的增加,单位的浓度增加,吸光度增加。
由图8,L1、L2、L3分别在不同极性溶剂中的发射光谱可知,随着溶剂极性的增加,它们的荧光强度逐渐减小,最大荧光发射波长逐渐红移。这是因为在极性溶剂中溶质.溶剂的相互作用强烈,处于松弛态的S1(第一激发态)相比于S0(基态)更容易被微环境所稳定,使得S1与S0之间的能级差减小,因此发射光谱红移[15]。由图 8 知,L1、L2、L3在THF溶剂中的最大发射波长基本都在510 nm处,荧光强度相差不大,以荧光素为参比测得的相对量子产率分别为0.76,0.48,0.37,随着n的增加发光效率降低,可能由于枝数的增加,增加了分子的空间位阻,非辐射跃迁的概率增大,不利于荧光的产生。

图5 L1~L3的红外吸收光谱

图6 L1~L3的红外吸收光谱
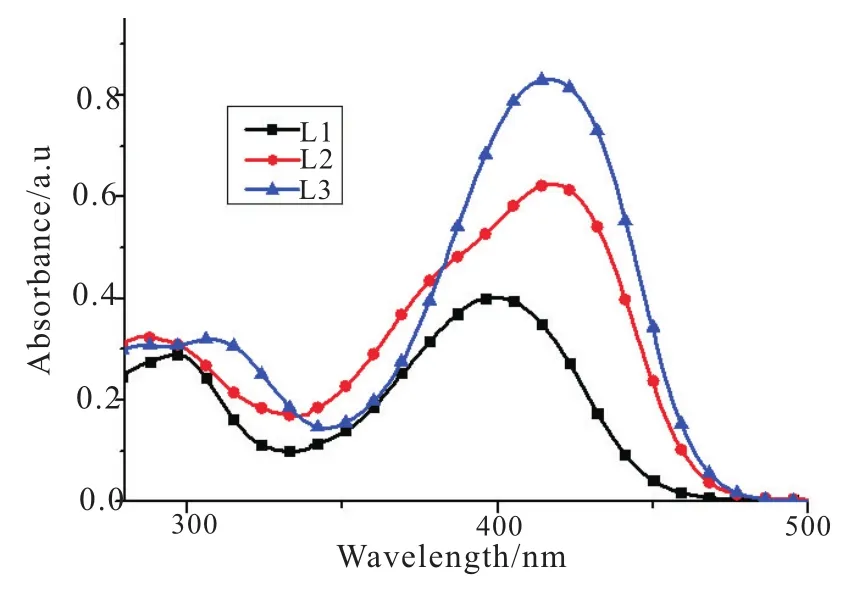
图7 L1~L3在THF溶剂中的紫外-可见吸收光谱
2.3 理论计算
为从理论上更深刻的理解D-(D'-A)n(n=1~3)型醛基化合物,n的变化对电子吸收光谱的影响,利用G09软件,采用TDDFT(PBE38/6-31G),计算了它们在THF中的的电子吸收光谱。结果如表1。计算值比实验值红移约20 nm。化合物理论计算的结果和实验值相当吻合,n=2时,吸收波长最大。由图10可知,低能带的吸收来自于HOMO→LUMO的跃迁。电子云从HOMO轨道的三苯胺基团,向LUMO轨道的醛基发生了转移。

图8 L1~L3在不同溶剂中的紫外-可见吸收光谱

表1 化合物L1~L3相关的理论和实验数据

图9 L1~L3在不同溶剂中的荧光光谱

图10 L1~L3最高占据分子轨道和最低空缺分子轨道图
3 小结
以卤代三苯胺、5-醛基-2-噻吩硼酸为原料,[1,1-双(二苯基磷)二茂铁]二氯化钯为催化剂,通过Suzuki偶联反应,合成了D-(D'-A)n型醛基化合物L1、L2、L3,研究了它们的光谱性质,探讨了随着n的增加,它们性质的在THF溶剂中的变化:在相同的浓度下,长波处的吸收强度随着n的增加,吸光强度增强,而最大发射波长和发光强度没太大变化,荧光量子产率随着n的增加而减弱。
猜你喜欢
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
设备管理与维修(2019年9期)2019-09-12
当代化工研究(2016年1期)2016-03-16
合成化学(2015年9期)2016-01-17
合成化学(2015年10期)2016-01-17
大连工业大学学报(2015年4期)2015-12-11
中国塑料(2015年7期)2015-10-14
应用化工(2014年12期)2014-08-16
应用化工(2014年9期)2014-08-10
