
基于特征图谱及指标成分优化新清毒饮颗粒的制备工艺
2024-03-05 06:53陆小梦古玉正刘瑞媚林银慧周珊宇何欣欣肖飞张军黄新安广州中医药大学青蒿研究中心广东广州50405广州中医药大学科技产业园有限公司广东广州50445广州中医药大学中药学院广东广州50006
中药新药与临床药理 2024年2期
陆小梦,古玉正,刘瑞媚,林银慧,周珊宇,何欣欣,肖飞,张军,黄新安(. 广州中医药大学青蒿研究中心,广东广州 50405;. 广州中医药大学科技产业园有限公司,广东广州 50445;. 广州中医药大学中药学院,广东广州 50006)
新清毒饮由国医大师邓铁涛师承团队治疗病毒性感冒的临床经验方[1]加减而来,由蒲公英、金银花、野菊花等17味中药组成,具有清热解毒、解表化湿、止咳平喘之功效,方中金银花、蒲公英等多味药材对流感病毒有良好的抑制活性,临床上用于由流感病毒等引起的上呼吸道感染等疾病[2-5]。为了方便临床使用,同时保证汤剂疗效,将其开发成颗粒剂。依据《中药新药质量标准研究技术指导原则(试行)》[6],中药复方制剂应建立其特征图谱并制定相应标准,并以相似度、相对峰面积等为检测指标控制其质量。中药特征图谱现已广泛用于中药提取工艺环节的质量控制、药材和制剂的整体评价[7-8],但未见将特征图谱与指标成分保留率相结合考察复方制剂制备工艺的研究。蒲公英为处方中的君药,其主要成分菊苣酸具有抗病毒、抗菌、抗炎、调节免疫等药理作用[9-11]。2020 年版《中国药典》增加了蒲公英中菊苣酸的含量测定。处方中甘草的主要成分甘草酸具有抗病毒、抗炎、抗过敏等作用[12-14]。中药复方制备过程中有效成分保留率易受煎煮温度、溶剂、料液比等的影响[15-17];在中试或大生产中,不稳定药效成分如金银花、蒲公英中绿原酸、菊苣酸等酚酸类成分经过相对长时间的提取、浓缩、干燥,保留率或有降低[18-20]。本研究拟建立新清毒饮的特征图谱与蒲公英中菊苣酸、甘草中甘草酸的含量测定方法,以追踪各工艺步骤中有效成分的保留情况,优化其制备工艺,并开展中试生产验证优选的工艺路线,关注并克服规模生产下不稳定药效成分的降解,制备与原方汤剂物质基础近似的颗粒剂,以期重现新清毒饮汤剂临床疗效。
1 仪器与材料
SECVRA225D-1CN 十万分之一电子天平、BSA224S-CW 万分之一电子天平(德国赛多利斯有限公司);LC-20AT 高效液相色谱仪(日本岛津公司);ZORBAX-SB-Phenyl色谱柱(4.6 mm×250 mm,5 μm,美国安捷伦公司);Symmertry®C18色谱柱(4.6 mm×250 mm,5 μm,美国Waters 公司);KQ-250DE 超声清洗仪(昆山市超声仪器有限公司);R205B 旋转蒸发仪(上海申胜生物科技有限公司);3000 L 多功能提取罐、1000 L 减压浓缩机组(江苏沙家浜化工设备有限公司);PS1000-NC 高速离心机(辽阳中联制药机械有限公司);ZKS-5型真空上料机(江阴市宏达分体设备有限公司);FZ200沸腾制粒机(浙江迦南科技股份有限公司);DXDK-80C 颗粒全自动包装机(珠海市天楹精密机械有限公司);甲醇、乙腈(色谱纯,德国Merck 公司);水为超纯水,其他试剂均为分析纯。
对照品菊苣酸(批号:111752-202105,纯度98.3%)、绿原酸(批号:110753-202119,纯度96.3%)、芦丁(批号:100080-202012,纯度92.2%)、甘草苷(批号:111610-201908,纯度95%)、木犀草苷(批号:111720-202111,纯度96.6%)、蒙花苷(批号:111528-201911,纯度98.5%)、补骨脂素(批号:110739-201918,纯度99.6%)、甘草酸铵(批号:110731-202021,纯度96.2%),中国食品药品检定研究院。处方饮片来源信息见表1。
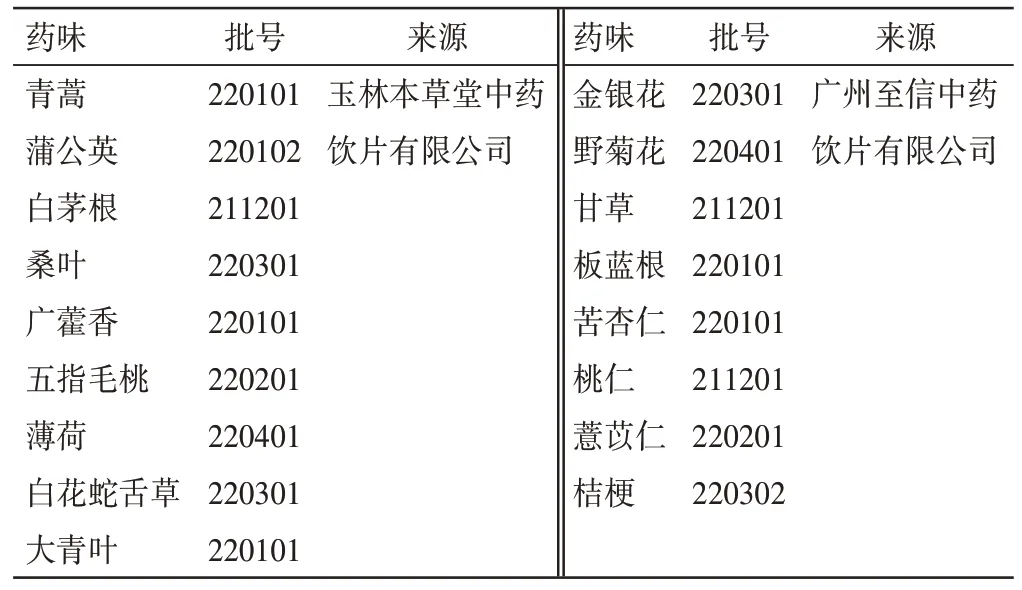
表1 新清毒饮药材来源信息Table 1 Source informations of Xinqingduyin
2 方法与结果
2.1 特征图谱测定方法的建立
2.1.1 色谱条件 色谱柱为ZORBAX SB-Phenyl(4.6 mm × 250 mm,5 μm);流动相:乙腈(A)-0.1%磷酸(B),梯度洗脱(见表2);检测波长:240 nm;流速:0.9 mL·min-1;柱温:30 ℃;进样量:10 μL。

表2 特征图谱测定的流动相洗脱梯度Table 2 Elution gradient of mobile phase used in HPLC characteristic spectra
2.1.2 混合对照品溶液的配制 取对照品适量,精密称定,加甲醇制成每1 mL含菊苣酸、绿原酸、芦丁、甘草苷、木犀草苷、蒙花苷、补骨脂素、甘草酸分别为73.436、394.400、7.328、0.912、1.602、0.966、6.135、8.070 μg的混合对照品溶液。
2.1.3 新清毒饮颗粒及阴性颗粒的制备 按处方比例称取17 味药材饮片,加8 倍量水煎煮3 次,每次1 h,煎煮3 次,过滤,合并滤液;取滤液80 ℃减压浓缩成浸膏(含饮片生药量2 g·g-1);取浸膏70 ℃干燥并粉碎成浸膏粉,以适量乳糖作辅料,制粒,即可得新清毒饮颗粒。按处方比例分别称取缺单味药材的饮片,同法制备新清毒饮阴性颗粒。
2.1.4 新清毒饮单味药材配方颗粒的制备 取单味药材的饮片50 g,同“2.1.3”项制备成单味药材配方颗粒。
2.1.5 供试品、阴性对照、单味药材样品溶液的配制取新清毒饮颗粒1 g,精密称定,置具塞锥形瓶中,精密加入50%甲醇溶液50 mL,密塞,称定质量,超声处理20 min,放至室温。再称定质量,用50%甲醇溶液补足减失的质量,摇匀,滤过,即得新清毒饮供试品。另取阴性颗粒、单味药材配方颗粒各1.0 g,同法制备阴性对照液、单味药材对照液。
2.1.6 特征图谱专属性试验与指认 取“2.1.2”“2.1.5”项下各溶液,按“2.1.1”项下方法进样检测,考察实验方法的适用性及专属性。特征图谱见图1。样品中各峰分离良好,峰形对称。通过阴性对照液、单味药材溶液与供试品溶液色谱比较,对色谱峰进行归属和定位,共标定16个峰。1号峰归属于白花蛇舌草,6、10 号峰归属于金银花,8、16 号峰归属于甘草,12号峰归属蒲公英,14号峰归属野菊花、桑叶,15号峰归属五指毛桃,2、3、4、5、7、9、11、13号色谱峰分别归属于多个药味共有峰。通过对照品对照,鉴别了5号峰为绿原酸,7号峰为芦丁,8、16号峰为甘草苷和甘草酸,9号峰为木犀草苷,12号峰为菊苣酸,14号峰为蒙花苷,15号峰为补骨脂素。
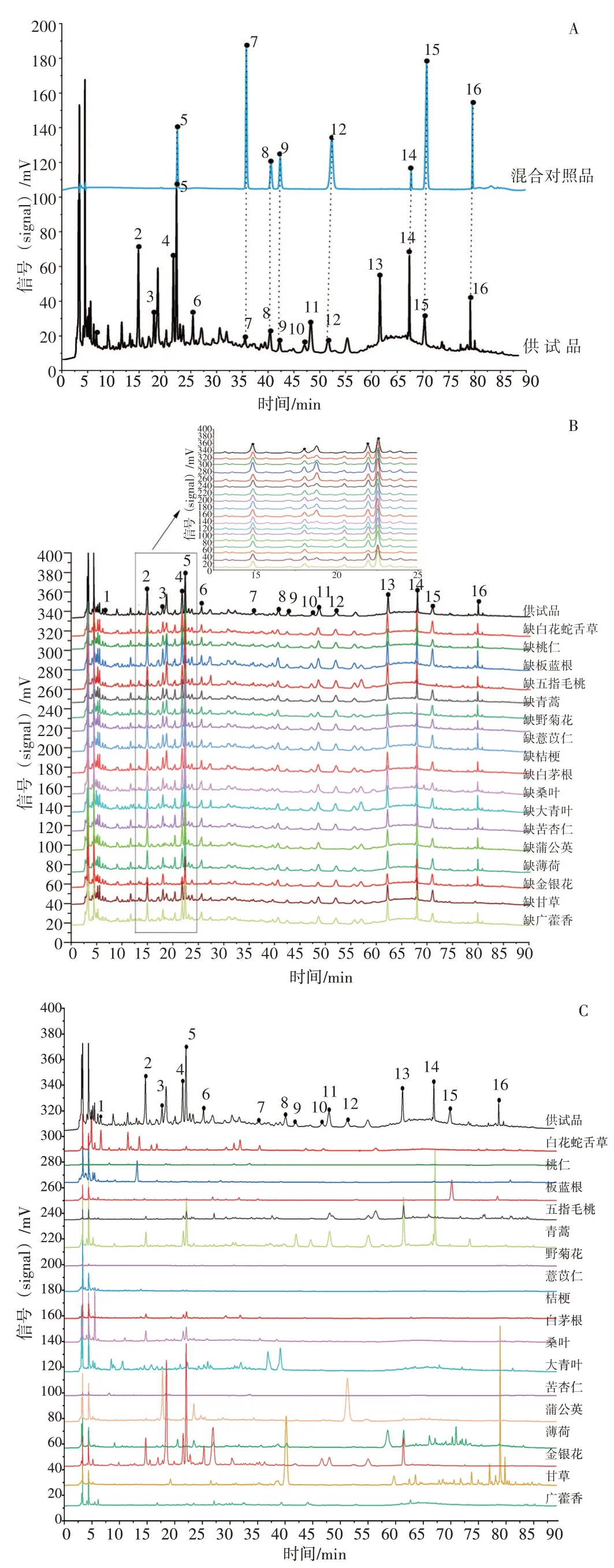
图1 新清毒饮颗粒高效液相色谱(HPLC)特征图谱Figure 1 HPLC fingerprint of Xinqingduyin Granules
2.1.7 精密度试验 精密吸取同一份供试品溶液,按“2.1.1”项下色谱条件连续进样6 次,记录色谱峰,以5 号峰(绿原酸)为参照峰(S),计算各峰相对保留时间和相对峰面积。结果显示,1~16 号峰的相对保留时间和相对峰面积的RSD 分别为0.01%~1.01%和0.15%~1.22%,表明仪器的精密度良好。
2.1.8 稳定性试验 取同一份供试品溶液,分别于0、3、6、12、18、24、36 h 进样测定,结果表明1~16 号峰的相对保留时间和相对峰面积的RSD 分别为0.01%~1.11%和0.12%~1.54%,表明供试品溶液在36 h内稳定性良好。
2.1.9 重复性试验 精密称取新清毒饮颗粒6 份,按“2.1.5”项制备供试品溶液,进样测定,以5号峰(绿原酸)为参照峰,结果显示1~16号峰的相对保留时间和相对峰面积的RSD 分别为0.01%~0.18%和0.29%~3.21%,表明该方法重复性较好。
2.2 含量测定方法
2.2.1 色谱条件 (1)菊苣酸:色谱柱为Agilent ZORBAX SB-Phenyl(4.6 mm × 250 mm,5 μm);流动相:乙腈(A)-0.1%磷酸溶液(B),梯度洗脱(见表3);流速:0.9 mL·min-1;柱温:30 ℃;进样量:10 μL;检测波长:327 nm;理论塔板数按菊苣酸峰计算应不低于5 000。

表3 菊苣酸含量测定的流动相洗脱梯度Table 3 Elution gradient of mobile phase in the content determination of chicory acid
(2)甘草酸:色谱柱为Symmetry®C18(4.6 mm×250 mm,5 μm);流动相:乙腈(A)-0.1%磷酸溶液(B),梯度洗脱(见表4);流速:1.0 mL·min-1;柱温:31 ℃;进样量:10 μL;检测波长:252 nm;理论塔板数按甘草酸峰计算应不低于5 000。

表4 甘草酸含量测定的流动相洗脱梯度Table 4 Elution gradient of mobile phase in the content determination of glycyrrhizic acid
2.2.2 对照品溶液的制备 取菊苣酸对照品适量,精密称定,加甲醇制成浓度为151.480 3 μg·mL-1的菊苣酸对照品溶液。同法制成浓度为471.441 2 μg·mL-1的甘草酸对照品溶液。
2.2.3 供试品及阴性对照溶液制备 新清毒饮颗粒供试品溶液、缺蒲公英阴性对照液和缺甘草阴性对照液制备方法同“2.1.3”项下。
2.2.4 专属性试验 精密吸取“2.2.2”“2.2.3”项下各个溶液,按“2.2.1”项下色谱条件进行测定。供试品溶液色谱图中,在与对照品溶液色谱相应位置上有相同保留时间的色谱峰,菊苣酸峰、甘草酸峰分离度良好,阴性无干扰。见图2。

图2 新清毒饮颗粒的高效液相色谱图Figure 2 HPLC chromatography of Xinqingduyin Granules
2.2.5 线性关系考察 取“2.2.2”项下菊苣酸对照品溶液,加甲醇稀释配制75.740 2、30.296 1、15.148 0、12.118 4、6.059 2、3.029 6 μg·mL-1的系列对照品浓度。同法,取甘草酸对照品溶液配制成188.576 5、94.288 2、37.715 3、18.857 6、9.428 8、4.714 4 μg·mL-1,依法进样测定,以浓度(X)为横坐标,峰面积(Y)为纵坐标,绘制标准曲线。菊苣酸线性方程为:Y=52 686.18X-28 106.94,r=1.000 0,表明菊苣酸在3.029 6~151.480 3 μg·mL-1范围内,线性关系良好。甘草酸线性方程为:Y=8 026.63X+509.10,r=0.999 8,表明甘草酸在4.714 4~471.441 2 μg·mL-1范围内,线性关系良好。
2.2.6 精密度试验 取同一份菊苣酸、甘草酸对照品溶液,按“2.2.1”项重复进样6 次测定,结果菊苣酸、甘草酸的峰面积的RSD 分别为0.92%、0.23%,表明精密度良好。
2.2.7 稳定性试验 取“2.2.3”项下同一份供试品溶液,分别于0、8、12、16、24 h 进样,结果菊苣酸的峰面积均值为477 754,RSD 为0.51%,甘草酸峰面积均值为120 273,RSD 为0.22%。结果表明供试品溶液24 h内稳定性良好。
2.2.8 重复性试验 取同一批新清毒饮颗粒6 份,按“2.2.3”项制备供试品溶液,依“2.2.1”项下方法测定。测得菊苣酸平均含量为0.246 1 mg·g-1,RSD 为0.92%,甘草酸平均含量为0.758 1 mg·g-1,RSD 为0.48%。结果表明本方法重复性良好。
2.2.9 加样回收测定 取已知含量的新清毒饮颗粒0.5 g,各6份,精密称定,分别加入与颗粒中菊苣酸含量、甘草酸含量相等的对照品溶液,按“2.2.3”项下方法制备供试品并进样测定,计算得菊苣酸、甘草酸回收率分别为99.77%、97.74%,RSD 分别为1.09%、2.79%。结果说明本方法可靠,结果准确。
2.3 工艺考察
2.3.1 提取工艺考察 按照临床汤剂应用经验,本试验采用水煎煮法。按照预实验结果,采用L9(34)正交试验设计,将加水量(A)、提取时间(B)、提取次数(C)作为影响因素,因素与水平见表5。按正交试验设计,制备S1~S9 号试验溶液,取试验溶液依“2.1”“2.2”项下方法测定特征图谱及指标成分含量,计算保留率。
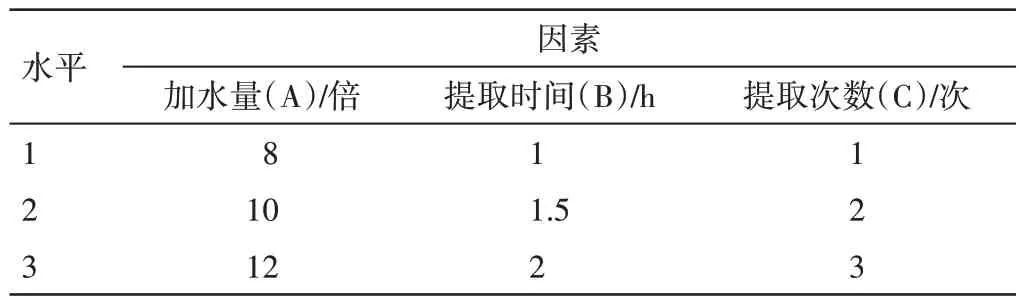
表5 新清毒饮颗粒的正交试验因素水平表Table 5 Design of factors and levels of orthogonal test for Xinqingduyin Granules
2.3.1.1 特征图谱结果分析 结果见图3。从S1~S9 号试验溶液图谱可见,1~16 号特征峰均可保留,表明加水量、提取时间、提取次数等因素改变对成分种类未造成影响;以1 号试验药液(S1)中5 号峰(绿原酸)为参照峰,计算相对峰面积。结果见表6。各峰的相对峰面积RSD 较大,即对成分含量有影响,故应以指标成分的保留率结果优化工艺参数。

图3 新清毒饮颗粒的正交试验特征图谱的叠加图Figure 3 Superimposed characteristic chromatogram of orthogonal test for Xinqingduyin Granules

表6 新清毒饮颗粒的正交试验特征图谱相对峰面积结果Table 6 Results of relative peak area of characteristic chromatograms in orthogonal experiment of Xinqingduyin Granules
2.3.1.2 以指标成分保留率为指标优化工艺参数 设置菊苣酸保留率(X)、甘草酸保留率(Y)的权重系数分别为0.6、0.4,综合评分(M)=(0.6X/菊苣酸最大的保留率+0.4Y/甘草酸最大的保留率)×100,对上述3个因素进行考察,优选较佳的提取工艺。试验结果见表7,方差分析见表8。

表7 新清毒饮颗粒的正交试验及结果Table 7 Orthogonal test and results of Xinqingduyin Granules

表8 新清毒饮颗粒的正交试验方差分析Table 8 Analysis of variance for orthogonal test of Xinqingduyin Granules
结果显示,各因素对试验结果的影响大小依次为:C>A>B,加水量(A)、提取时间(B)均无显著性影响,提取次数(C)对试验结果有显著性影响。为节约成本,缩短生产周期,保证有效成分的保留率,结合方差分析结果,拟采取最佳提取工艺:A1B1C3,即加8倍量水,提取3次,每次1 h。
2.3.2 浓缩工艺考察
2.3.2.1 浓缩方式考察 以“2.3.1”项下确定的提取工艺制备新清毒饮提取液,作为浓缩工艺对照样品。准确量取2 500 mL提取液各2份,分别考察常压、减压浓缩成含饮片量2 g·g-1浸膏。取浓缩液,同“2.1”项下方法进样测定特征图谱,以对照样品的特征图谱中5 号峰为S 峰,计算各峰相对峰面积;依次将样品的色谱图导入“中药色谱指纹图谱相似度评价系统(2012 年版)”软件,以对照样品色谱图为参照,对样品色谱峰进行多点校正和Mark 峰匹配,计算相似度。同时,按“2.2”项下方法测定甘草酸、菊苣酸含量,计算保留率。
从特征图谱可见,两种浓缩方式均能保留1~16 号特征峰(见图4);常压、减压浓缩特征图谱的相似度结果分别为0.963、0.999,均大于0.9,说明两种方式未对成分种类造成显著性影响;相同生药量下,6、7 号等特征峰的相对峰面积差异较大(见表9),表明不同浓缩方式对含量有一定影响,故以指标成分保留率为指标进一步优化工艺参数。

图4 新清毒饮颗粒不同浓缩方式的特征图谱叠加图Figure 4 Superimposed characteristic chromatograms of Xinqingduyin Granules with different concentrative methods
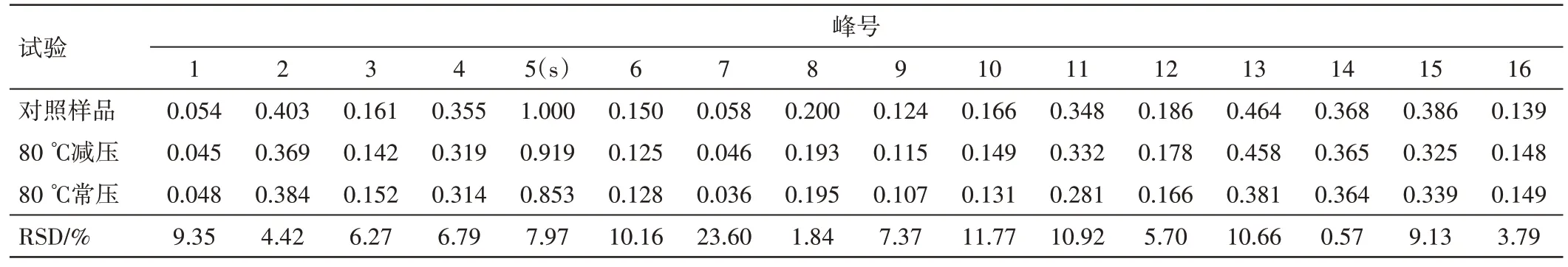
表9 新清毒饮颗粒不同浓缩方式特征图谱的相对峰面积Table 9 The relative peak area of characteristic peaks of Xinqingduyin Granules with different concentrative methods
与对照样品对比,减压浓缩对试验结果的影响较小(见表10),且浓缩时间短,结合相似度结果,最终选择减压浓缩。

表10 新清毒饮颗粒不同浓缩方式指标成分保留率结果Table 10 Results of retention rates of index components in Xinqingduyin Granules with different concentrative methods
2.3.2.2 浓缩温度考察 按“浓缩方式”操作,准确量取提取液2 500 mL,各2 份,考察不同浓缩温度(60~90 ℃)对特征图谱及指标成分保留率的影响。
从特征图谱可见,不同浓缩温度均能保留1~16 号特征峰(见图5);浓缩温度60~90 ℃特征图谱的相似度结果均为0.999,说明不同浓缩温度对成分种类未造成显著性影响;不同浓缩温度的特征图谱中,1、6、7 号峰的相对峰面积差异略大,余峰差异不大(见表11),表明温度对样品的成分含量仍有一定的影响,故应进一步考察浓缩温度对指标成分保留率的影响。
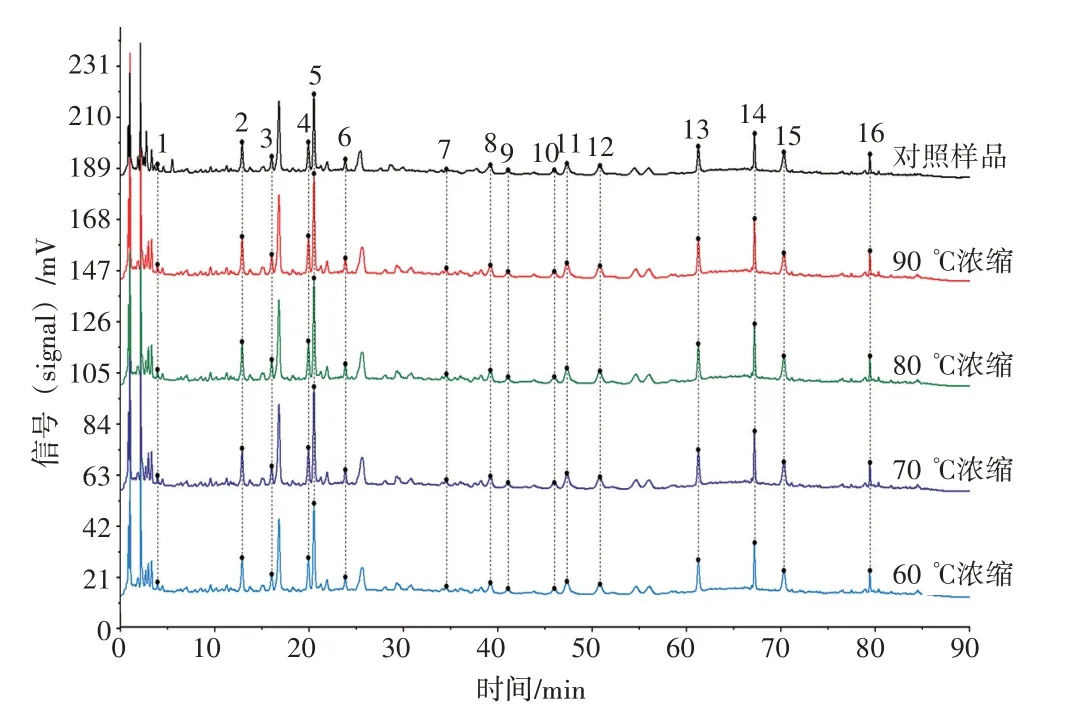
图5 新清毒饮颗粒的不同浓缩温度的特征图谱叠加图Figure 5 Superimposed characteristic chromatograms of Xinqingduyin Granules under different concentrative temperatures

表11 新清毒饮颗粒不同浓缩温度的特征图谱相对峰面积Table 11 The relative peak area of characteristic chromatograms of Xinqingduyin Granules under different concentrative temperatures
由综合评分结果可知,随着浓缩温度上升,综合评分逐渐降低,但无显著性差异(见表12)。为缩短生产周期,保证成分保留率,选择80 ℃减压浓缩作为最佳工艺。
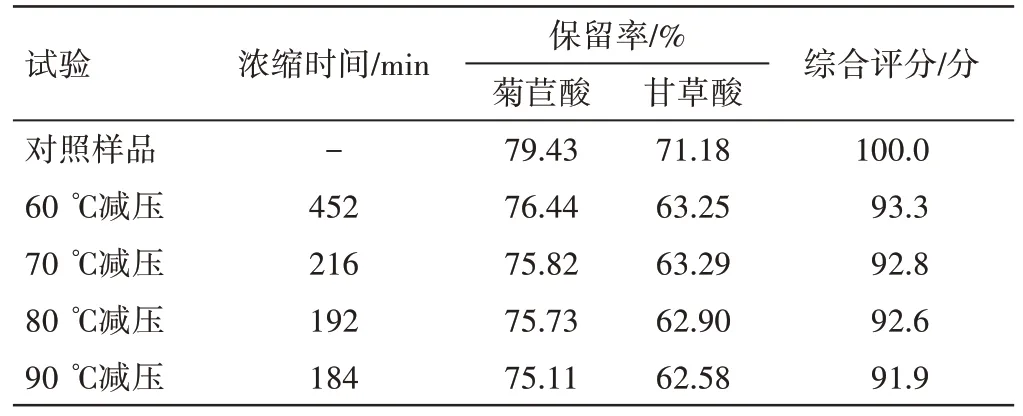
表12 新清毒饮颗粒不同浓缩温度的指标成分保留率结果Table 12 Results of retention rates of index components in Xinqingduyin Granules under different concentrative temperatures
2.3.3 成型工艺考察 按优选工艺,取“2.3.2”项下提取液,于80 ℃减压浓缩成含饮片量2 g·g-1的浸膏样品;称取浸膏于70 ℃减压干燥制成浸膏粉,备用。
2.3.3.1 辅料种类筛选 取浸膏粉48 g(相当于181 g饮片量),各2份,分别与糊精、乳糖、可溶性淀粉、甘露醇4 种辅料1∶1 混匀,喷入75%乙醇适量,混匀,制成软材,过筛,制成颗粒。考察软材状态、制粒难度、成型率、吸湿率等指标以优选辅料种类。
结果显示,乳糖成型率最高且吸湿性最小,优于可溶性淀粉,甘露醇制得颗粒质地较硬,颜色不均匀,糊精所得颗粒口感差(见表13),故优选乳糖为辅料。

表13 新清毒饮颗粒制粒辅料的筛选Table 13 Screening of excipients for granulation of Xinqingduyin Granules
2.3.3.2 辅料用量配比考察 取浸膏粉48 g,各2 份,分别与不同乳糖配比制成颗粒,以颗粒成型性、吸湿性、休止角等为指标考察辅料用量。
结果显示,当浸膏粉与乳糖用量配比为1∶1.14或1.2∶1.5时,颗粒成型率高,休止角小,吸湿性小(见表14)。为减少服用量,故选择浸膏粉∶乳糖的比例为1∶1.14。
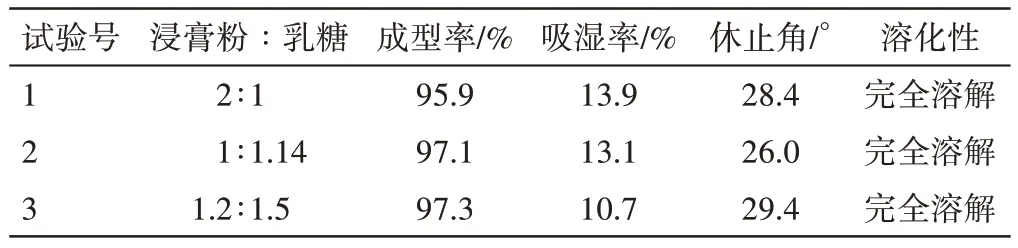
表14 新清毒饮颗粒辅料用量的考察结果Table 14 Investigation result of excipient dosage of Xinqingduyin Granules
2.3.3.3 矫味剂考察 按上处方配比制成颗粒,经口尝具有一定苦涩味,故在相关研究[21-22]基础上优选甜菊素作为本品矫味剂,并通过不同用量的优选考察,当甜菊素用量为成品质量的0.5%时,口感得到改善。故确定本品甜菊素用量为成品质量的0.5%。
2.4 中试工艺及样品测定 根据实验室优选的制备工艺,按处方称取17 味中药,共181 kg,共3 份,加8 倍量水,煎煮3 次,每次1 h,药液滤过。合并滤液,80 ℃下减压浓缩成浸膏(相对密度1.10~1.20,60~70 ℃),加入成品质量0.5 %的甜菊糖苷溶液(浓度约为10 %,m/V),以适量乳糖(浸膏粉∶乳糖=1∶1.14)作辅料,一步制粒,分装(每袋10 g),即得。中试工艺过程数据的相关结果见表15。依法测定中试提取液、浸膏、颗粒中菊苣酸、甘草酸的含量,计算保留率,并测定各个药液的特征图谱,以中试提取液(T601)为参照图谱,计算相似度,并以5号峰(绿原酸)为参照峰(S),计算各个峰的相对峰面积。结果见图6和表16、表17。
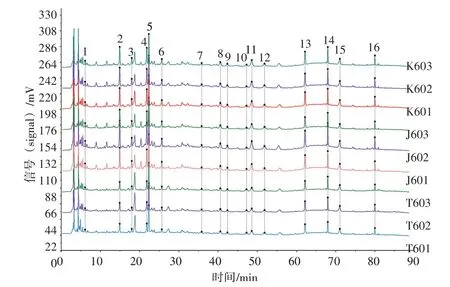
图6 新清毒饮颗粒中试工艺提取液-浸膏-颗粒的特征图谱叠加图Figure 6 Superimposed characteristic chromatograms of pilotprocess extraction-extractum-granule of Xinqingduyin Granules

表15 新清毒饮颗粒中试工艺的相关结果Table 15 The corresponding results of pilot process steps of Xinqingduyin Granules
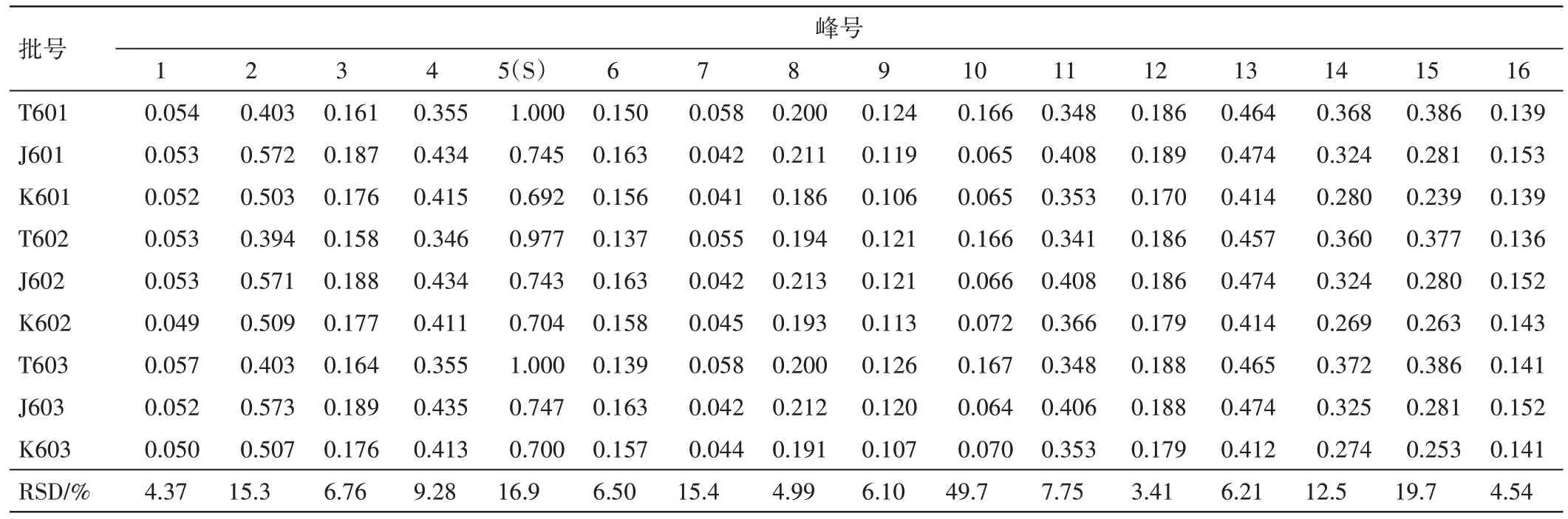
表16 新清毒饮颗粒中试工艺提取液-浸膏-颗粒的特征图谱相对峰面积结果Table 16 The relative peak area of characteristic peaks of pilot-process extraction-extractum-granule of Xinqingduyin Granules

表17 新清毒饮颗粒中试工艺提取液-浸膏-颗粒的指标成分保留率结果Table 17 The results of the retention rate of index component for pilot-process extraction-extractum-granule of Xinqingduyin Granules
2.4.1 中试工艺相关数据结果分析
2.4.1.1 特征图谱 1~16 号峰在提取液-浸膏-颗粒的工艺过程中(见图6),以T601色谱图为参照,中试浸膏、颗粒的特征图谱相似度分别为0.970、0.973,表明该工艺未对药液成分种类造成显著影响;同一工艺步骤中,提取液、浸膏、颗粒各批次样品之间的相似度均为0.999,提示本中试工艺重复性良好。与提取液比较,颗粒剂中5、7、10、14、15 号峰相对峰面积有所减少,其余峰稳定,表明该工艺对成分的含量稍有影响。见表16。
2.4.1.2 指标成分保留率 从提取液到颗粒的中试工艺过程中(见表17),菊苣酸保留率由(76.11±0.99)%降低至(54.56±1.63)%,甘草酸由(77.96±1.79)%降低至(54.96±1.08)%,表明中试工艺过程成分保留率下降幅度较大。
2.4.2 中试颗粒与同批饮片汤剂比较 汤剂的制备[23]:按处方比例称取17药味饮片共181 g,共3份。加水没过药面2 cm,浸泡30 min(广藿香、青蒿、薄荷除外),武火煮开后转文火煎25 min。加入单独浸泡30 min 的广藿香、青蒿、薄荷,继续煎煮5 min,过滤。药渣加水没过药面2 cm,二次煎煮30 min。过滤,合并滤液,即得新清毒饮汤剂。依法测定汤剂的特征图谱及指标成分含量,计算保留率,与中试样品比较分析。
2.4.2.1 特征图谱分析 依次将3 批汤剂样品的色谱图导入“中药色谱指纹图谱相似度评价系统软件(2012 年版)”,以T1 号样品色谱图为参照,对3 批样品色谱峰进行多点校正和Mark 峰匹配,生成对照色谱图(R0)。同法,依次生成3批中试提取液对照色谱(R1)、浸膏对照色谱(R2)、颗粒对照色谱(R3),取R0~R3 的色谱图导入上述软件系统,以R1 为参照图谱,计算相似度。结果显示,从汤剂至成分颗粒均可见1~16 号特征峰(见图7)。R0~R3 相似度结果为1.000、0.975、0.968、0.971,均大于0.96,证明经过中试生产的新清毒饮颗粒与汤剂主要成分相似。
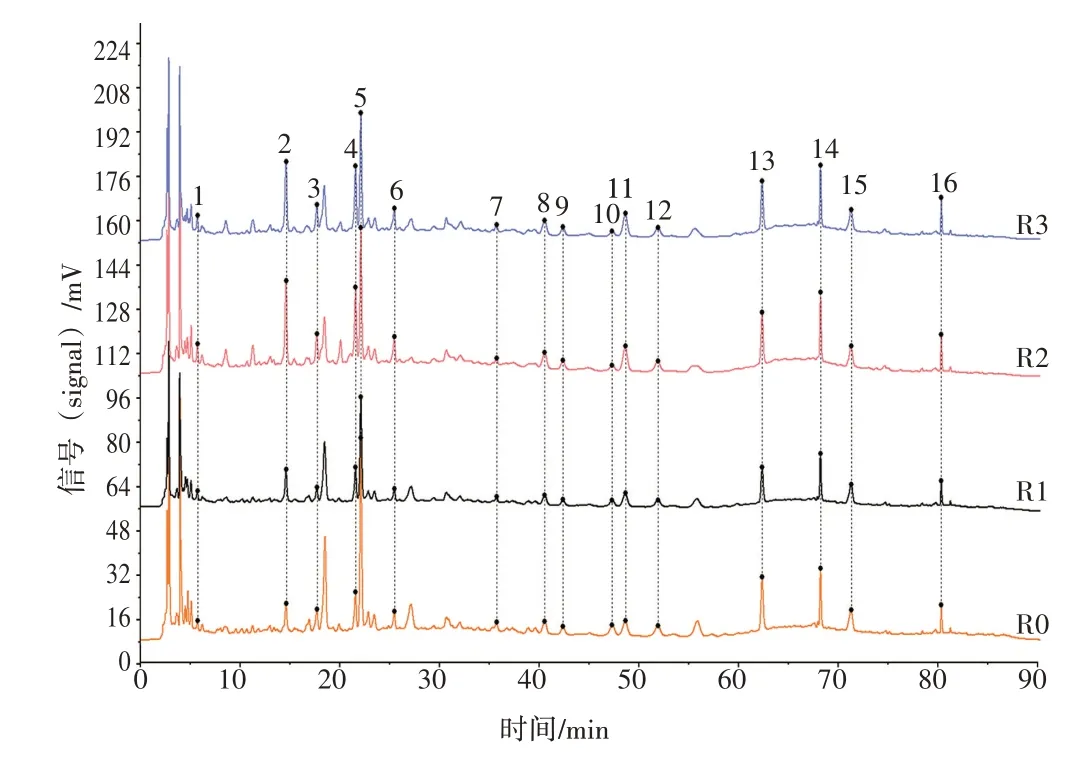
图7 新清毒饮汤剂-中试颗粒的对照图谱叠加图Figure 7 Superimposed reference chromatogram of Xinqingduyin decoction-pilot-process granule
2.4.2.2 指标成分保留率结果 汤剂中菊苣酸、甘草的保留率为(52.88±4.00)%、(45.21±1.71)%,与中试成品颗粒中菊苣酸、甘草酸保留率相比,虽然中试规模生产成品颗粒成分保留率呈现较大幅度降低,但结果均高于汤剂中的成分保留率(见表17和表18),结合特征图谱,进一步证明新清毒饮颗粒工艺路线的可靠性。

表18 与中试同批饮片制备的新清毒饮汤剂的指标成分保留率Table 18 The retention rate of index component of Xinqingduyin decoction prepared from the same batch of decoction pieces as the pilot-batch
3 讨论
3.1 新清毒饮汤剂、小试与中试样品比较 本研究进行中试验证前,通过放大投料量,按照优选工艺制备3 批新清毒饮小试颗粒,取制备过程中的提取液、浸膏、颗粒样品测定特征图谱及含量,计算指标成分保留率。以提取液样品的色谱图为参照,浸膏、颗粒的色谱图相似度均大于0.9。对小试、中试工艺的样品及汤剂中指标成分保留率进行比较(见表19),发现中试样品中菊苣酸、甘草酸的保留率显著下降,成品颗粒中菊苣酸保留率显著低于小试工艺样品的保留率,可能是大规模生产往往加热时间较长,导致菊苣酸受热分解,使其保留率下降[16]。中试提取、浓缩过程中甘草酸保留率较高,但在成品颗粒中显著下降,其原因可能与一步制粒过程中的泵进药液速度、进风温度、雾化压力、喷射速度等影响因素有关[24-25]。但是,新清毒饮颗粒中菊苣酸、甘草酸保留率仍显著高于新清毒饮汤剂。
表19 新清毒饮颗粒小试、中试工艺与传统汤剂指标成分的保留率比较[(±s),%]Table 19 Comparison of retention rate of the index component among small-pilot-trail process and traditional decoction of Xinqingduyin Granules[(±s),%]

表19 新清毒饮颗粒小试、中试工艺与传统汤剂指标成分的保留率比较[(±s),%]Table 19 Comparison of retention rate of the index component among small-pilot-trail process and traditional decoction of Xinqingduyin Granules[(±s),%]
样品提取液浸膏颗粒小试工艺保留率菊苣酸78.89±3.59 74.66±3.41 67.60±1.74甘草酸58.47±2.81 57.19±3.23 55.42±5.25中试工艺保留率菊苣酸76.11±0.99 62.81±2.55 54.56±1.63甘草酸77.96±1.79 72.51±1.05 54.96±1.08汤剂保留率菊苣酸52.88±4.00甘草酸45.21±1.71
3.2 特征图谱结果分析 中试样品色谱图及相对峰面积结果显示,新清毒饮颗粒工艺过程样品与对照图谱相似度均大于0.9,表明该工艺过程中,样品主要成分种类未发生显著变化。5 号峰(绿原酸)、7 号峰(芦丁)、10 号峰(归属金银花的未知成分)、14 号峰(蒙花苷)、15 号峰(补骨脂素)的相对峰面积RSD 较大,表明这些成分含量存在较大的差异。另外,图7中保留时间18.5 min处的未标识峰,在成品颗粒中峰面积显著减少,因此后期研究应关注该成分是否为有效成分,并进一步关注热不稳定有效成分的降解,通过优化工艺,提高有效成分保留率,以期制备成药效成分高、质量均一的颗粒剂。
本研究在小试试验中对整个制备工艺进行考察,确定了新清毒饮初步的生产工艺,利用中试规模加以验证,对评价工艺放大的稳定性具有重要的意义;所建立的特征图谱方法适用于新清毒饮颗粒各生产流程的质量评价,为不同工艺过程产物的质量控制提供了参考;中试工艺制得新清毒饮颗粒剂与同批次饮片制得汤剂的比较显示,特征图谱相似性高,菊苣酸、甘草酸保留率高,可为中药复方制剂工艺研究提供参考。
猜你喜欢
西北药学杂志(2023年4期)2023-07-27
中医药导报(2021年4期)2021-11-22
天然产物研究与开发(2019年1期)2019-03-01
世界科学技术-中医药现代化(2018年9期)2019-01-29
中成药(2017年5期)2017-06-13
中国药业(2014年21期)2014-05-26
中国药业(2014年4期)2014-05-09
中国刑警学院学报(2014年4期)2014-04-27
云南畜牧兽医(2014年2期)2014-02-28
新疆农垦科技(2014年2期)2014-02-28
