
褐煤与水微观相互作用的分子机制:多尺度分子模拟
2024-03-21 04:51吴玉花李荣花高红凤朱美霖刘彩珠吴建波白红存
洁净煤技术 2024年2期
吴玉花,李荣花,张 茜,高红凤,朱美霖,刘彩珠,吴建波,张 慧,白红存
(1.宁夏大学 省部共建煤炭高效利用与绿色化工国家重点实验室,宁夏 银川 750021;2.宁夏大学 化学化工学院,宁夏 银川 750021;3.宁夏医科大学 基础医学院,宁夏 银川 750004)
0 引 言
煤炭高效清洁利用是我国高质量发展的重大现实需求。煤炭资源作为世界上最丰富的化石能源之一,自人类进入工业和现代化社会以来始终占据重要地位。在我国,煤炭是关键的能源和化工原料。据国家统计局公布的历年国民经济和社会发展统计公报,我国能源消费中煤炭始终占50%以上。就煤炭资源分布和利用现状而言,低阶煤占比极大。褐煤是重要的典型低阶煤资源[1-2],化学反应活性较强,但目前仍存在一些问题,如不易储存和运输,暴露在空气中易发生风化等。此外,相较于无烟煤和烟煤,褐煤含水量更高而含碳量更低,其在燃烧过程中需消耗更多热量来蒸发水分,这使得褐煤燃烧效率相对较低,同时产生更多烟尘、SOx和NOx等污染物。然而,优质煤炭资源不断消耗,褐煤已成为我国煤炭开采利用的主要煤种之一,在煤炭产量中占比达12.7%。因此,针对褐煤开展相关研究对煤炭高效清洁利用具有重要现实意义。
煤与水的表面相互作用是煤炭清洁高效利用领域的重要科学问题之一[3-5]。在煤炭开采、加工和利用中,煤与水的表面相互作用在不同过程和场景下广泛存在。煤炭勘探过程中,存在地下水流经煤层并溶解煤层中有毒物质的可能。煤炭开采时,矿井水和地下水不仅影响开采速度,还可能导致各种安全问题。煤炭储运阶段,煤与水的相互作用也不容忽视,是引发煤自燃的重要因素。煤炭加工转化过程中,水的存在会对燃烧效率造成重要影响;而煤气化、煤制氢等过程中水作为原料直接参与化学反应。此外,煤与水的表面相互作用对煤炭加工过程污染物的生成和排放也有重要影响。低阶煤,特别是褐煤,因其独特的地质形成过程,在物理和化学特性上表现出较高的内水和多孔结构[1,6]。这些特性使褐煤与水的相互作用在微观尺度上变得复杂且具有特殊意义。可见,厘清煤与水的表面相互作用,尤其是微观相互作用的分子机制,对于褐煤等低阶煤勘测、开采、储运和综合利用十分必要。
煤与水的表面相互作用是一个复杂的物理化学过程,受多种因素影响,包括煤的种类、含水量、粒度、温度、压力等。研究煤与水的表面相互作用,需从试验和理论计算两个方面进行[7-9]。试验上,相关学者利用X射线衍射、扫描电子显微镜、热重分析等方法,研究了煤表面的水分吸附、溶解、沉淀、胶结等典型过程,目前已开展了较多研究,在微纳尺度和微颗粒尺度取得了不少进展。KELEMEN等[10]发现,褐煤具有类胶体结构特性,在干燥、脱水和吸附过程中,煤的孔隙结构会收缩和膨胀;当水分被蒸发时,孔结构发生不可逆性收缩。水从褐煤脱除时主要通过颗粒固结和结构坍塌而排出,且并非所有含水的孔都会发生水分脱除[11]。此外,还通过分子热力学和表面物理化学方法,对煤吸附水的微观机理进行理论分析。研究发现,水分子被煤吸收是水分子与煤表面相互吸引所致[12],且煤分子与水的相互作用力主要是范德华力和氢键[13]。这些研究为煤-水相互作用的微观机理提供了理论依据。然而,目前对于煤和水相互作用的试验研究仍主要停留在微纳尺度的宏观现象和规律描述的解读阶段,尚缺乏分子尺度的系统刻画,需要开展化学精确尺度的机理研究。另一方面,计算机软硬件不断发展,利用量子化学、分子动力学模拟等分子模拟方法在理论上研究煤与水的表面相互作用成为可能。量子化学计算结果表明,煤分子表面能更稳定地吸附CO2和H2O,其中非共价键相互作用占主导地位[14]。在氢键协同效应下,随着水分子数量增大,体系中的氢键总能随之增大[15],褐煤中各种含氧官能团与水的相互作用能由大到小依次为羧基、酚羟基、羰基、脂羟基、醚[16]。根据氢键供体的不同,褐煤与水形成的氢键可分为2类,即HO—H…O和C—H…OH2。前者由于相对更短的作用距离通常比后者的相互作用更强[16]。通过密度泛函理论计算可以给出煤与水分子相互作用的能量、势能曲面、构象能等。相关研究还阐述由于氢键、π—π堆积及其他非共价相互作用的存在,使褐煤分子中持有较高的含水量[17-18]。也有研究利用分子动力学模拟考察煤/添加剂/水悬浮体系相互作用[19-21],为煤-水体系中分散剂/表面活性剂的分子行为提供微观解释。
虽然水与低阶煤中含氧官能团的相互作用受到一定关注,但二者相互作用机理仍不清楚。采用分子动力学研究煤与水的相互作用仍有待深入。这些工作对于理解褐煤等低阶煤炭资源的勘测、开采、储运和综合利用过程中煤-水相互作用相当重要。由此引发两个问题:首先,褐煤分子中各种杂原子官能团对水分子吸附的稳定性和能量的标准是什么?第二,煤炭勘测、开采、储运和利用过程中煤-水相互作用均发生在非真空的现实环境中,此时水分子大量存在,而密度泛函理论(Density Functional Theory, DFT)计算通常给出真空条件下的计算结果,模拟条件与现实情况不符。基于分子动力学模拟现实情况下煤与大量水分子相互作用的微观分子机制将如何?
笔者基于多尺度分子模拟方法探究褐煤与水的微观相互作用。通过量子化学BLYP-D3和CCSD(T)方法对褐煤与单分子水的相互作用进行研究,重点考察典型褐煤结构中杂原子官能团对水的吸附,通过分析体系能量、相互作用区域和类型定性和定量研究褐煤与水之间相互作用的微观物理本质。还利用分子动力学模拟对多个褐煤分子与大量水的相互作用进行研究,考察统计平均近似下大量煤-水相互作用,揭示其组装行为和相互作用机制。在分析褐煤与水单分子相互作用时使用量子化学计算,可以给出基于量子力学的精确结果,如电子结构、结合能量、化学键和静电势分布等。但受限于计算成本,只能对相对较小的体系进行处理。当考虑多个褐煤分子与大量水分子相互作用时,系统内原子数急剧增加(>10万),这超过了量子化学处理的能力,因此采用分子动力学进行处理。本工作结合两种方法的各自优势,从不同角度深入研究褐煤与水的相互作用。计算模拟的尺度从电子结构、原子位置、分子构象到聚集颗粒,跨越多个层次和尺度。这些工作的开展对理解煤与水微观相互作用的分子机制,实现煤炭高效清洁利用具有指导意义。
1 模型及计算方法
1.1 计算模型
煤根据变质程度可分为褐煤、烟煤、无烟煤。煤的分子结构主要包含芳香片段和脂肪支链[22-24]。褐煤属于典型低阶煤,变质程度和煤化程度低,其分子结构特点是芳环缩合程度低且含大量含氧和含氮的杂原子官能团。参考已有文献构建C24H33O7N分子模型作为褐煤典型结构模型用于研究褐煤与水的相互作用。需要指出的是,该褐煤分子结构模型参考了WENDER[25]、TROMP等[26]提出的具有代表性的褐煤结构,其中包含羧基、酚羟基、脂羟基、氨基和醚键等典型杂原子官能团。类似分子结构模型也已成功用于褐煤结构与反应性的分子模拟研究[16,25-27]。
褐煤分子与单分子水相互作用存在多种吸附位点。静电势(Electrostatic Potential, ESP)用来研究复杂体系中分子间的相互作用及分子反应性[28]。负静电势一般与具有更大电负性的原子联系紧密。因此ESP最小值在范德华表面的分布常被用来确定亲电攻击的有利位置。相反,正静电势常被用来确定亲核试剂进攻的有利位置。分子模拟研究中采用的褐煤分子模型C24H33O7N 及静电势分布如图1所示(原子标识:浅蓝C;灰色H;红色O;深蓝N)。由图1(b)可知,在褐煤分子结构静电势中,蓝区和红区是最活跃的位点,分别更易发生亲核反应和亲电反应。红区和蓝区明显存在于杂原子官能化位置。可见,褐煤分子中含杂原子基团的位置活性强,更易发生反应。这为褐煤中各基团对水分的吸附提供了有力支撑。

图1 分子模拟研究中采用的褐煤分子模型C24H33O7N 及静电势分布
褐煤分子与单分子水相互作用不仅存在多种吸附位点,即使同一吸附位点,还可能存在多种吸附构象[29]。因此,进行构型搜索是使用分子模拟方法研究分子结合的关键。这里采用系统式搜索法实现不同吸附构象的局域极小考察,优于常规的人为搭建有限构象的方法。系统式搜索法允许分子的某些二面角按指定规则旋转并将产生的结构作为优化的初始结构。使用MOLCLUS程序[30]中GENMER工具,灵活性高且可控性强,根据试验需求选择不同的单体种类和数量,调整发射方向和移动方式,这些优点可获得更多样化的结构。利用GENMER将水分子从褐煤分子的官能团中心原子位置向任意方向发射。在发射过程中,单体水分子不断运动,形成有效结构。为获得更多有效结构,利用褐煤分子中各种含氧官能团实现对水分子的高效吸附。通过该方法可充分探索不同官能团和水分子间的相互作用方式,从而更好地理解其之间的化学性质和结构特征。针对褐煤与水的相互作用,每个吸附位点考察20种初始结构,总计获得超300种初始结构提交结构优化和能量计算,最后筛选出不同吸附位点的15种低能量吸附构象,如图2所示(原子标示:浅蓝C;灰色H;红色O;深蓝N)。这些结构中包括水分子在入NH2处吸附3种,—OCH3和—OH处吸附各5种及—COOH处吸附2种。

图2 褐煤分子与水单分子相互作用的15种低能量吸附构象
1.2 量子化学计算细节
使用量子化学计算考察褐煤对水的吸附机理,主要采用基于DFT的自洽场分子轨道方法。单分子模型复合物的几何构型优化使用施加色散校正的BLYP-D3方法在def2-TZVP基组水平上完成。为定量描述水与煤分子的结合强弱,复合体系的能量(结合能Eb、变形能Edef以及反应能Einter)也使用相同方法和基组水平上借助Gaussian16程序计算完成。此外,还借助ORCA程序采用基于波函数的耦合簇CCSD(T)方法在def2-TZVP基组水平上计算褐煤分子与水复合物体系的能量,并将结果与DFT中BLYP-D3方法计算结果进行对比。CCSD(T)方法是目前量子化学领域高精度计算方法之一,其计算结果是量子化学中公认的“黄金标准”[31-32]。使用CCSD(T)深入刻画褐煤与水单分子相互作用的物理本质,进一步采用能量分解分析(Energy Decomposition Analysis,EDA)方案将复合物体系的相互作用能分解成4部分:Pauli排斥能(EPauli)、色散校正能(Edisp)、静电相互作用能(Eelestat) 和轨道相互作用能(Eorb)。EDA计算使用BLYP-D3/TZP方法和基组借助ADF平台完成。需要指出的是,这里EDA计算也使用了相同的BLYP-D3方法,但与之前的能量计算使用了不同基组。其主要原因在于目前支持EDA的程序非常少,而可以执行EDA计算的ADF程序仅支持诸如TZP类型的Slater基组,而非def2-TZVP类型的Gaussian基组。在量子化学计算中,采用不同的程序和方法是一种较普遍使用且被认同的策略[16,27,32-33]。
1.3 分子动力学模拟计算细节
为揭示褐煤分子与大量水的相互作用,采用分子动力学模拟方法研究不同数量的煤分子在大量水中的组装行为和作用机制。考虑周期性边界条件下单胞中所含煤分子为10、20、40、60、80、100和200。选择GROMOS54A7力场作为分子动力学模拟(Molecular Dynamics,MD)的基本参数,MD步长设置为2 fs。采用动力学模拟LINCS算法,在粒子运动时将所有键视为完整约束条件。在298.15 K、正则系综(Canonical Ensemble,NVT)条件下进行计算模拟。对于静电相互作用,使用粒子网格方法计算,实空间截止长度为1.1 nm。使用Lennard-Jones模型进行分子间相互作用模拟,截止距离为0.9~1.1 nm。MD计算采用基于TIP3P水模型的显示溶剂处理方法以便考虑复杂体系的能量变化。在MD模拟中,先对系统进行能量最小化,再进行100 ps初步模拟,最后执行20 000 ps下优化模拟。MD计算使用GROMACS[34]软件完成,并使用程序自带工具和分子可视化程序(Visual Molecular Dynamics,VMD)分析模拟的轨迹文件。体系能量包括非键能和成键能。成键能为键角弯曲能、键伸缩能、反转能和旋转能。非键能为分子间和分子内的非键相互作用,可分为2类:静电相互作用与范德华相互作用。静电作用主要是长程作用力,短程部分考虑了库仑排斥相互作用。而范德华作用主要是短程作用力,长程部分考虑了色散校正。
2 结果与讨论
2.1 褐煤与水单分子相互作用的量子化学分析
首先分析褐煤与水单分子相互作用稳定吸附构象的几何结构特征。针对褐煤与水单分子相互作用,采用系统式搜索法筛选出的局域极小构象经DFT方法全优化获得的结构如图2所示。褐煤与水单分子相互作用在—NH2官能团结合位点处筛选出3种不同的水分子吸附构型。在NH2-A中含氨基的链状结构与水分子中的氢垂直吸附,作用距离0.191 7 nm,而在NH2—C中则为0.233 8 nm平行吸附。在NH2-B中,H2O与—NH2以0.177 6 nm作用的同时,还与羧基(—COOH)中的H进行有效吸附(作用距离0.170 nm)。在—OCH3处吸附水分子筛选出5种结构模型。在OCH3-A中,H2O位于与—O—相连苯环的上方,呈现H…π相互作用。在OCH3-B中,H2O中的一个H朝向—O—,距离为0.194 5 nm。在OCH3-D中,H2O中的H与—OCH3中O距离为0.203 9 nm;OCH3-E中,H2O中的H与醚基中O的距离为0.202 8 nm,而另一H靠近苯环上方。在OCH3—C中,H2O中H在与煤中O形成氢键外,水中O还与临近的煤中H以0.205 0 nm有效吸附。—OH处吸附水分子筛选出5种构型。在OH-A中,H2O中的H与—OH的距离为0.194 2 nm。OH-B模型中,H2O中的O与—OH中H距离为0.210 3 nm。OH—C中,H2O中的H与—OH中O距离为0.201 4 nm。在OH-D构型中,H2O位于OH侧的上方,H2O中的H朝向苯环,H2O与苯环距离约0.254 nm,是典型的H…π相互作用。在OH-E结构模型中,—OH中O与H2O中的H距离为0.197 4 nm。在—COOH结合位点,筛选出2种合理构型。在COOH-A中,H2O中的H和O分别与—COOH中的O、H相互作用,其距离分别为0.217 4和0.184 4 nm。COOH-B中,H2O位于苯环上方,与—COOH相距0.199 6 nm。上述相互作用距离均在典型的氢键作用范围以内[8-9,14,35]。
利用独立梯度模型(Independent Gradient Model,IGM)[36-37],可以形象地反映褐煤和水的相互作用。褐煤与水分子之间不存在共价键,相互作用属于非共价类的弱作用力。IGM结果中颜色愈深,相互作用强度愈高。褐煤与水单分子相互作用的IGM分析如图3所示(浅蓝C;灰色H;红色O;深蓝N),褐煤与水之间的有效作用区域呈蓝色和绿色。因此,褐煤与水分子之间非共价相互作用主要包括氢键(蓝色)和范德华作用(绿色)。其中,氢键颜色更深,作用强度大于范德华作用,是主要作用力。根据键临界点(Bond Critical Point,BCP)处电子密度估计的氢键键能及该键的形成情况见表1。在醚基处形成的第1种吸附构象(OCH3-A),存在弱的氢键(O—H…π),其作用力仅5.796 kJ/mol,第2种吸附构象(OCH3-B)中氢键O—H…O─C的距离较近(0.194 nm),因而其强度可达19.908 kJ/mol。在OCH3—C形成2个强度为15.96 kJ/mol的氢键,在OCH3-D中形成1个强度为7.56 kJ/mol的氢键,在OCH3-E中形成2个强度为7.56 kJ/mol的氢键。水分子在褐煤的第1个—OH处将形成3种吸附构象,在官能团OH-A、OH-B和OH-C三种构型中,分别形成1个、2个和1个氢键。水分子在褐煤结构中第2个—OH处拥有2种吸附构象,分别形成1个氢键,其中OH-D具有较小的氢键强度(5.107 kJ/mol)。在COOH官能团中有2种不同的水吸附构象, COOH-A中形成2个作用强度均为29.736 kJ/mol的氢键,作用距离分别为0.217和0.184 nm,COOH-B中形成1个作用强度为17.052 kJ/mol的氢键且作用距离为0.199 nm。在氨基处形成3种不同的水分子吸附构象,其中在第1个构象(NH2-A)中形成1个强度为28.812 kJ/mol的(O—H…N─C)的氢键。在第2个构象(NH2-B)中形成2个强度均为41.454 kJ/mol的氢键(H—O…H─O和O—H…N─C)。最后1个吸附构象(NH2-C)中形成一个强度为8.4 kJ/mol的H—O…H─N氢键,且作用距离较长(0.234 nm)。因此,氢键的形成是影响褐煤-水复合系统稳定性的关键因素[8,38]。
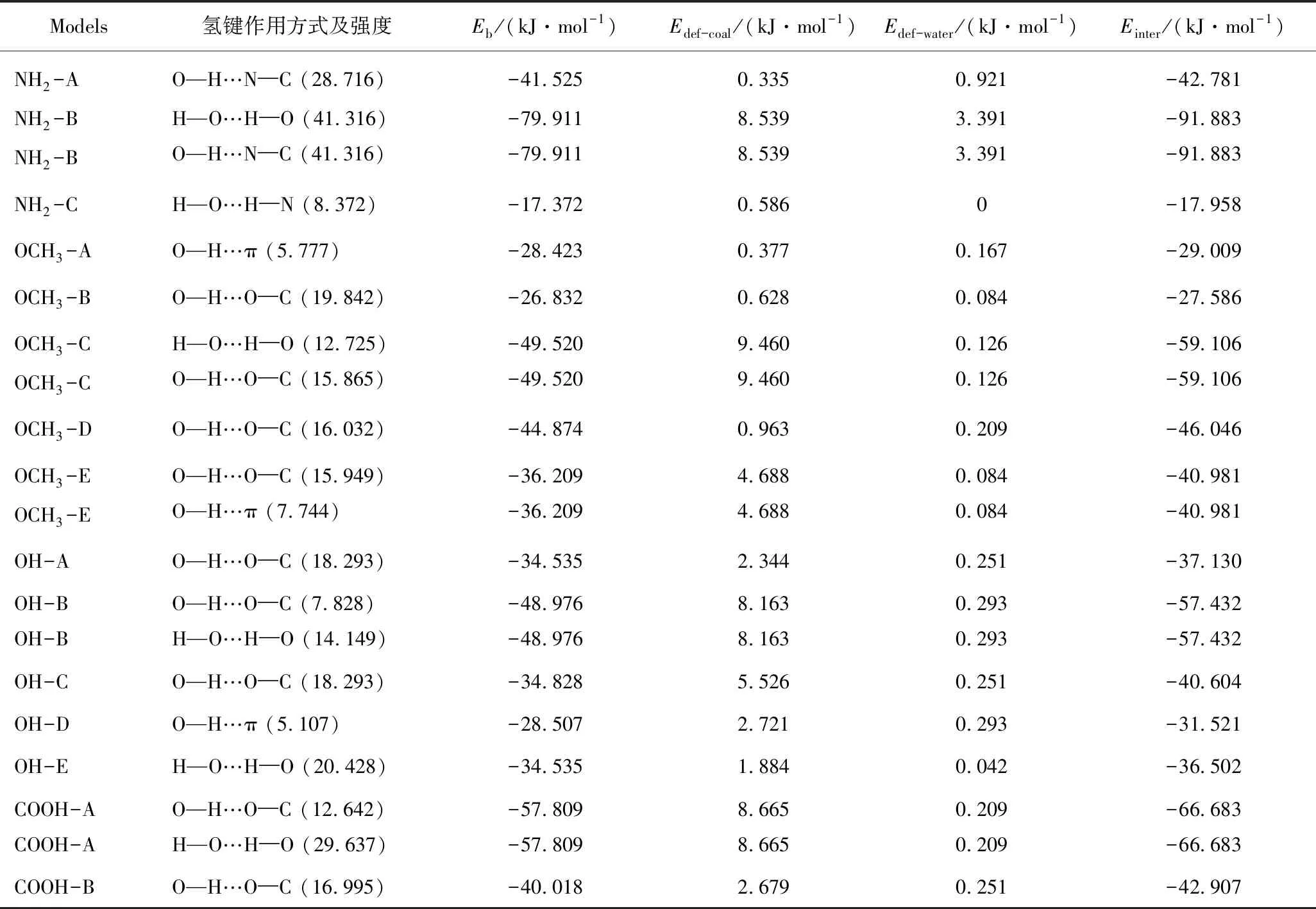
表1 稳定吸附构型中氢键作用方式、强度,及结合能Eb,变形能Edef和反应能Einter

图3 褐煤与水单分子相互作用的IGM分析
对褐煤和水分子间的相互作用进行定量研究。从能量的角度出发,研究褐煤-水复合体系中的氢键、范德华作用力、单体间的结合等因素对其稳定性的影响。在与结构优化相同的计算水平下,得出吸附过程的反应能(Einter),两单体的变形能(Edef-coal和Edef-water)及结合能(Eb),计算结果见表1。
最优单体之间结合所放出的能量为结合能,以下为相应的计算公式:
Eb=Ecoal+water-Ecoal-opt-Ewater-opt。
(1)
其中,Ecoal+water为煤-水复合体系通过充分优化后所得的能量;Ecoal-opt为褐煤分子的最优结构能量;Ewater-opt为水分子的最优结构的能量。值得注意的是,吸附过程中所有的结合能(Eb)均为负,因此从能量方面分析,有助于褐煤中不同官能团对水的吸附。H2O与褐煤中不同官能团的结合是有利的。在氨基处形成的3种吸附构象,NH2-B是吸附水的最佳位点,其结合能为-79.911 kJ/mol,是最稳定的吸附构象。与其他两种构象相比,NH2-B中存在双氢键,这说明褐煤分子对水的吸附作用由氢键作用引起。对于OCH3-A和OCH3-B结构,其结合能分别为-28.423和-26.832 kJ/mol。OCH3-A的复合体系中存在1个作用强度为5.777 kJ/mol的氢键。OCH3-B复合体系中存在1个作用强度为19.842 kJ/mol的氢键。可见2个体系所形成的氢键较弱。但OCH3-A中有很大区域的弱相互作用。这进一步证实了氢键、范德华力对褐煤-水作用的重要影响[8-9,38]。OCH3-C、OCH3-D和OCH3-E是褐煤中另一个—OCH3与水分子的吸附构型,在OCH3-C存在2个作用强度分别为12.725和15.865 kJ/mol的氢键。由于相对强的氢键作用,OCH3-C的结合能达到-49.520 kJ/mol。OCH3-D中不仅存在1个氢键,还存在大区域的弱相互作用,可能由于以上原因使得OCH3-D的结合能为-44.874 kJ/mol。OCH3-E中存在2个作用强度分别为15.949和7.744 kJ/mol的氢键,其结合能为-36.209 kJ/mol。由于OH-A和OH-C均形成1个作用强度为18.293 kJ/mol的氢键,使得OH-A的结合能为-34.535 kJ/mol,OH-C的结合能为-34.828 kJ/mol。由于OH-B形成2个作用强度分别为7.828和14.149 kJ/mol的氢键以及大区域的弱相互作用,使得OH-B的结合能高于OH-A和OH-C,达到-48.976 kJ/mol。对于水分子在褐煤分子中另1个OH官能团处的吸附,OH-D中存在1个作用强度为5.107 kJ/mol的氢键,结合能为-28.507 kJ/mol。OH-E存在1个作用强度为20.428 kJ/mol的氢键,其结合能为-34.535 kJ/mol。对于水分子在褐煤分子中—COOH官能团处的吸附,由于COOH-A中形成2个氢键,因此其结合能为-57.809 kJ/mol,而COOH-B的结合能为-40.018 kJ/mol。综上所述,褐煤中官能团—COOH、—OH、—OCH3以及—NH2吸附水分子的最佳位点分别对应COOH-A、OH-B、OCH3-C以及NH2-B。拥有最大结合能的为NH2-B,其次为COOH-A。
变形能为褐煤和水分子单体在形成褐煤-水吸附系统前后的差值,计算可由式(2)、(3)表示:
Edef-coal=Ecoal-Ecoal -opt,
(2)
Edef-water=Ewater-Ewater-opt。
(3)
其中,Edef-coal和Edef-water分别为褐煤分子和水分子的变形能;Ecoal和Ewater分别为复合体充分优化后与产生形变的褐煤和水分子单体能量,区别于Ecoal -opt和Ewater-opt分别对应褐煤和水单分子优化后的最低能量。表1给出了计算的变形能结果,可知变形能均为正,表明褐煤和水分子单体在吸附时,需进行结构变形。水分子的变形能量在0~3.391 kJ/mol。较小的Edef-water表示H2O发生较小的结构形变,甚至大部分情况下可忽略。较大的Edef-coal说明相对水分子,褐煤的变形更明显。褐煤分子中—NH2官能团结合位点处的NH2-A吸附构象拥有最小的变形能,仅为0.335 kJ/mol, OCH3-C吸附构象拥有最大的褐煤分子变形能,达到9.460 kJ/mol。当水分子被吸收后,褐煤分子基本上仍保留原来的形态,没有明显的构象变化。
分析褐煤与水分子相互作用的反应能Einter。分子间的相互作用一般可划分为2个阶段:① 分子间相互靠近时会发生形变;② 分子在形变后相结合[29,32]。反应能指形变发生后褐煤分子与水分子结合时释放的能量。在吸附过程中,反应能是能量变化的主要因素,可将其视为结合能和形变能之和。Einter可用式(4)计算:
Einter=Ecoal+water-Ecoal-Ewater。
(4)
由表1可知所有Einter均为负值,在热力学方面为褐煤-水复杂系统的形成提供理论依据。该结论与Eb相似。褐煤中各官能团处进行水分子吸附时,Einter绝对值在17.958 ~ 91.883 kJ/mol。其中,NH2-C中最小,而NH2-B中最大。由于Einter=Eb-Edef(Eb为结合能,Edef为变形能),而变形能数值较小,因此优势构象主要取决于褐煤与水相互作用的反应能。对于Einter而言,不同官能团处吸附水分子时反应能绝对值递减的排序依次为:氨基>羧基>脂肪醚基>脂羟基>酚羟基>芳香醚基。
将高精度量子化学计算结果与DFT中BLYP-D3计算结果进行对比分析。借助ORCA程序使用CCSD(T)/def2-TZVP方法和基组对褐煤与水单分子相互作用体系的Eb及Einter进行高精度计算,确保BLYP-D3/def2-TZVP计算的有效及可靠。图4为CCSD(T)和BLYP-D3的计算结果。根据CCSD(T)计算,结合能Eb绝对值在10.80~57.641 kJ/mol,NH2-C最小,NH2-B最大,这与BLYP-D3计算一致。
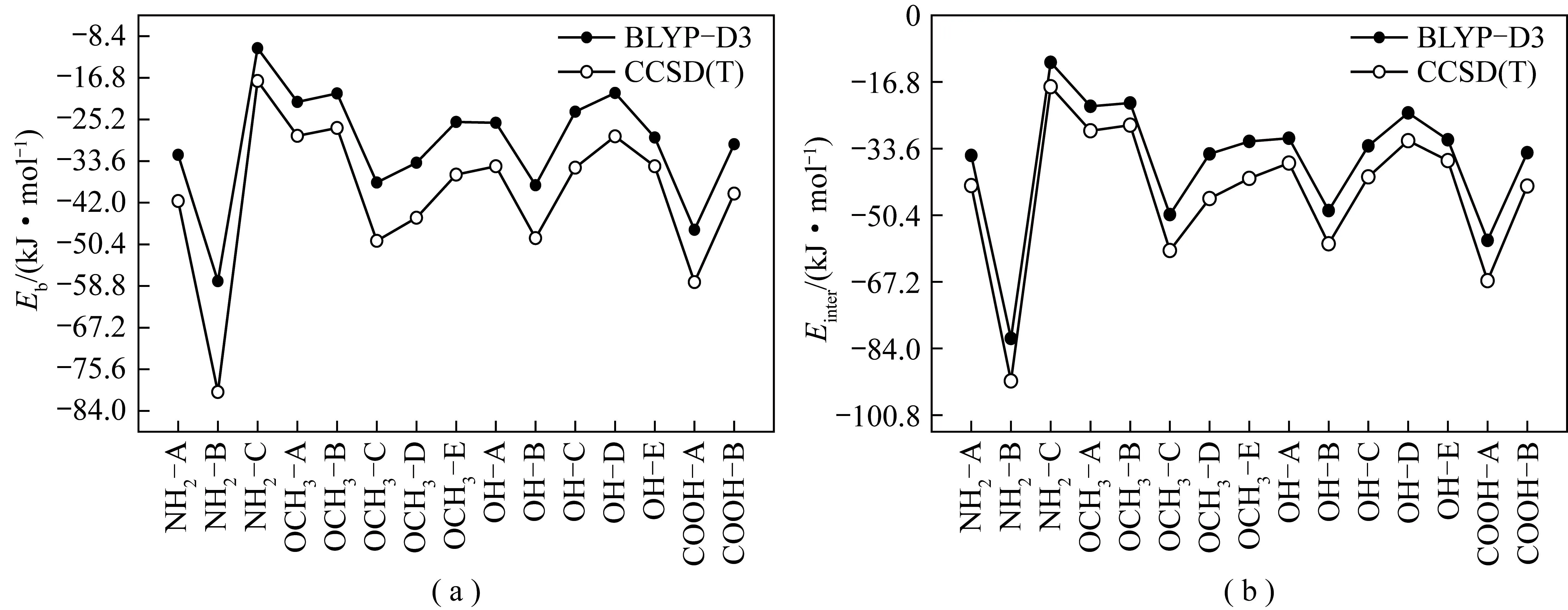
图4 BLYP-D3和CCSD(T)方法计算获得的Eb和Einter
对比发现,CCSD(T)计算值比BLYP-D3略负,但二者表现为相同趋势的近似平行线,其差值主要是CCSD(T)和BLYP-D3两种不同量子化学计算方法的系统误差所致。对于CCSD(T)方法计算得到的Einter,其绝对值在11.80 ~ 81.21 kJ/mol,均比BLYP-D3的绝对值更大。然而对比BLYP-D3结果,二者的相对误差仅为6.153~11.22 kJ/mol。综上,CCSD(T)和BLYP-D3相比,二者的计算结果一致性良好[39]。这为BLYP-D3方法研究煤-水相互作用的可靠性和有效性提供了证据。
通过EDA对褐煤与水单分子相互作用的物理本质进行定量解析。通过上述褐煤与水相互作用能量的计算结果分析可知,褐煤与水2个单体在吸附中起主要作用的是Einter。Einter反映了吸附的效果和难易程度。因此,有必要对其进行深入讨论。采用EDA方法,将反应能分为4种类型的物理相互作用部分[29,32,39]:
Einter=Eelestat+EPauli+Eorb+Edisp。
(5)
其中,Einter为总相互作用能;EPauli为Pauli排斥能;Eelestat为静电相互作用能;Edisp为色散校正能;Eorb为轨道相互作用能。使用BLYP-D3泛函在ADF软件TZP方法水平上计算得到的反应能与Gaussian16程序Def2-TZVP水平上计算的数值差仅为3.223~8.832 kJ/mol,在允许范围内,故计算结果具有可比性。首先讨论EPauli,即Pauli排斥能。在复合体系中,EPauli均为正值,表示Pauli排斥能对煤-水吸附产生消极影响。其中,NH2-B的EPauli值最大,为170.035 kJ/mol,而NH2-C的EPauli最小,为21.683 kJ/mol。此外,EPauli更小的情况下,Einter也更负。使Einter更负就需克服更多EPauli。这表明在Einter更负的体系中,Eelestat、Eorb和Edisp的作用更为重要。
以下对Eorb、Edisp和Eelestat分别进行分析。Eorb,即轨道相互作用能,其通常主要涉及HOMO-LUMO间的相互作用。褐煤与水相互作用的能量分解分析见表2。由表2可知,Eorb结果均为负值。这意味着轨道相互作用对稳定复合体系有积极的影响。Eorb在总吸引作用(包括Eorb、Edisp和Eelestat)中的贡献比例为20.21%~39.31%,因此,Eorb对于含水褐煤的稳定性有重要作用。Eorb绝对值在7.493~100.464 kJ/mol,在NH2-C中绝对值最小而在NH2-B中绝对值最大。Edisp数值均为负值,表示Edisp有助于两个单体的吸附作用。其绝对值在10.758~29.721 kJ/mol,占总吸引作用的6.40%~34.92%。可知在多种相互作用力中,Edisp的贡献并不一定比Eorb显著。Eelestat主要指2个单体间的电子密度相互叠加引起的能量降低,对于复合物的稳定具有积极作用。由Eelestat结果可知,其绝对值为14.735~138.766 kJ/mol,在NH2-B中绝对值最大,而在NH2-C中最小。一般认为,静电力在氢键作用中起支配作用。褐煤中各杂原子官能团与水分子相互作用体系中,由于氢键的存在,Eelestat对总吸引力的贡献度约50%。因此,多种相互作用力中,Eelestat贡献最大,是稳定煤-水相互作用的核心因素。

表2 褐煤与水相互作用的能量分解分析
2.2 褐煤与大量水相互作用的分子动力学模拟分析
通过构建不同分子数(10、20、40、60、80、100、200)褐煤的水溶液体系,研究褐煤分子在水中的分子动力学行为。使用GROMOS54A7力场,步长2 fs,对其进行能量极小化、以及100和20 000 ps模拟。包含10、60和200个褐煤分子的初始模型,以及在水溶液中完成动力学模拟的模型如图5所示。可知,由于整个系统中的各个分子间和分子内均存在相互作用,使得褐煤分子在大量水溶剂中模拟发生团聚,且出现组装成为褐煤颗粒结构的现象。这与之前研究煤的单分子结构组装成为不同尺寸颗粒的结果类似[40]。因此,讨论煤-水分子间及分子内相互作用的能量非常重要。

图5 褐煤与大量水相互作用的分子动力学模拟前后的煤分子结构变化
选择褐煤分子和大量水作为研究对象,进行分子动力学模拟的能量计算,得到表3中体系能量组成的计算结果。首先分析成键相互作用。成键相互作用主要包括G96Angle、G96Bond、Proper Dih.以及Improper Dih.等4种相互作用。在成键项中,各项值均为正数,且主要的作用形式为G96Angle和G96Bond。当褐煤分子数目增加时,成键各项也随之单调增加。平均每分子褐煤的成键项数值为219.87 kJ/mol。

表3 分子动力学模拟不同数量褐煤分子与大量水分子相互作用的能量分析
对于非成键相互作用能量,Coulomb-14为褐煤分子与片段水的静电相互作用,LJ-14为褐煤分子与片段水的范德华作用,Coul. recip.为静电相互作用中的长程相互作用,Coulomb (SR)为静电相互作用中的短程相互作用,Disper. corr.为褐煤分子和水的范德华长程相互作用,LJ(SR)为褐煤分子和水的范德华短程相互作用。由表3可知,随褐煤分子数量增加,LJ-14,Coulomb-14,LJ(SR),Coul. recip.的绝对值增加。然而每个褐煤分子数保持相对稳定。具体而言,平均每分子褐煤的LJ-14为32.316 kJ/mol,Coulomb-14为-539.743 kJ/mol,LJ(SR)为15 288.319 kJ/mol,Coul. recip.为399.596 kJ/mol。当褐煤分子数为10时,Coulomb(SR)为-934 159.313 kJ/mol和Disper. corr.为-6 304.576 kJ/mol。然而,当褐煤分子数增至20时,Disper. corr.的绝对值瞬间增大,而Coulomb(SR)的绝对值瞬间减小。当褐煤分子数为20~200时,Coulomb(SR)和Disper. corr.的平均值相对稳定,即每分子褐煤的Coulomb(SR)为628.528 kJ/mol和Disper. corr.为-93 186.513 kJ/mol。值得注意的是,当褐煤分子数为10~200时,每褐煤分子的非键相互作用能近似,为-78 667.624 kJ/mol。需要强调的是,LJ(SR)、LJ-14和Coul. recip.为正值,而各非成键项之和为负值,因此这三者不利于多分子水在褐煤上的吸附。褐煤分子与水分子主要以静电作用结合,与之前使用EDA方法分析煤与水单分子相互作用的结论一致。
通过模拟不同数量的煤分子与大量水分子相互作用,考察系统的能量变化、氢键数量和溶剂可及表面积(Solvent-Accessible Surface Area, SASA),进一步研究褐煤分子与水分子的动力学性质。结果如图6所示,通过MD的100 ps进行研究,从能量变化角度对系统进行分析。由于事先已进行分子模拟中的能量极小化,因此系统能量迅速趋于平衡。当褐煤分子数目为10时,体系能量为-659 002.24 kJ/mol,势能为-785 354.64 kJ/mol,SASA为0.493 4 nm2;当分子数目为20时体系能量为-1 308 807.14 kJ/mol,势能为-1 559 546.85 kJ/mol,溶剂可及表面积为0.959 3 nm2;分子数目为40时体系能量为-2 603 937.53 kJ/mol,势能为-3 103 066.97 kJ/mol,溶剂可及表面积为1.696 9 nm2;分子数目为60时体系能量为-3 957 694.51 kJ/mol,势能为-4 716 158.41 kJ/mol,SASA为2.549 3 nm2;分子数目为80时体系能量为-5 233 291.56 kJ/mol,势能为-6 236 689.14 kJ/mol,SASA为3.546 6 nm2;当分子数目增至100时体系能量为-6 641 299.01 kJ/mol,势能为-7 914 469.24 kJ/mol,SASA为4.031 8 nm2;当分子数目最后为200时,能量达-13 173 391.19 kJ/mol,势能达-15 699 166.90 kJ/mol,SASA达8.498 7 nm2。以上体系能量和势能均为模拟50~100 ps的平均值,可知褐煤分子数与褐煤-水体系能量呈正相关,且每个褐煤分子平均能量为-65 728.36 kJ/mol。该体系的势能具有类似能量的变化规律,当体系趋于平衡后,该势能与褐煤分子数呈正比。每个褐煤分子的势能为-78 327.53 kJ/mol。与能量和势能相比,SASA稳定的平衡时间较长,随系统规模的增大,SASA也逐渐增大,但对于每个褐煤分子而言,SASA趋近于定值,即0.044 2 nm2。综上,通过分析能量、势能和SASA的变化趋势,得到以下结论:系统能量和势能与褐煤分子的数量呈正比,而每个褐煤分子的SASA趋于定值。结果对进一步研究煤与水的相互作用及煤的溶解过程具有重要的参考价值。

图6 MD模拟不同数量褐煤分子与大量水相互作用体系的能量、势能、溶剂可几表面积和氢键的变化
此外,水分子与褐煤中官能团之间形成的氢键是影响褐煤含水量的重要因素。通过动力学模拟,在多分子水吸附过程中,发现褐煤中逐渐形成的氢键数目与褐煤的数量有关,二者呈正比。在模拟中,褐煤数量不断增多时所形成氢键数目分别为56.84、121.20、222.12、350.55、457.13、548.48和1 134.70,可知每个褐煤分子所形成的氢键数量为固定值。经计算,每个褐煤分子关联5.72个氢键。该数值与图1(b)中褐煤单分子结构静电势显著分布区域的数量大致相当。
由上述分析可知,在多水分子吸附过程中能量、势能、SASA和氢键的统计结果与煤分子中官能团关系密切[15,40]。
3 结 论
1) 采用系统式搜索法筛选结合DFT结构优化获得了褐煤与水单分子相互作用不同吸附位点的15种局域极小构象。借助独立梯度模型考察分子间的相互作用,揭示了褐煤与水两分子间的相互作用形式主要为范德华作用和氢键。水分子吸附在褐煤不同位点时,通过不同作用方式的氢键形成了稳定的吸附构象,对褐煤分子与水复合体系的稳定起到重要作用。总结了稳定吸附作用的几何结构特征。
2) 从吸附结合能、形变能和反应能的能量计算多角度定量阐明了煤-水相互作用体系的稳定性规律。研究表明,相比形变能而言,反应能对复合物体系的稳定性具有更显著的决定性作用。采用EDA方法将煤-水分子间相互作用分解为静电作用,Pauli排斥作用,轨道作用,以及色散校正4种物理成分。定量分析结果表明,由于氢键的存在,静电作用能对总吸引力的占比约50%,是稳定煤-水相互作用的最主要因素。
3)借助MD模拟揭示了不同数量褐煤分子与大量溶剂水分子相互作用的组装行为和演化特征。褐煤分子结构在大量水中出现一定程度的团聚,呈现出组装为煤颗粒的结构。当褐煤分子数增加时,成键各项也随之单调比例增加。褐煤和大量水分子之间的作用主要以静电形式存在。通过MD模拟发现,褐煤分子数量直接影响该系统的能量变化、氢键数量和溶剂可及表面积。
猜你喜欢
大学物理(2022年9期)2022-09-28
选煤技术(2022年1期)2022-04-19
云南化工(2021年6期)2021-12-21
物理通报(2020年7期)2020-07-01
温州大学学报(自然科学版)(2016年1期)2016-10-27
浙江大学学报(工学版)(2016年2期)2016-06-05
原子与分子物理学报(2015年3期)2015-11-24
文理导航(2015年26期)2015-09-29
应用化工(2014年7期)2014-08-09
无机化学学报(2014年5期)2014-02-28
