
活性金属Ni d电荷密度对Ni2P/Al2O3催化剂菲加氢性能的影响
2024-03-21 04:53荆洁颖赵泽敏
洁净煤技术 2024年2期
荆洁颖,李 泽,赵泽敏,张 雨
(太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024)
0 引 言
高温煤焦油中菲含量高,占煤焦油总量4%~6%,与航空燃料的碳数一致(C9~C15)[1-2]。催化加氢是将菲转化为燃料的重要途径,将菲进行深度加氢处理可得密度大、热值高的全氢菲,热氧化稳定性和低温性能优良,可用作高能量密度燃料,用于航空航天[3-8]。这可提升菲的利用率,且产品附加值高,氢耗量低,符合国家要求。然而,菲加氢过程中的中间产物会与菲竞争表面吸附位点,不利于催化剂对菲吸附活化,使催化剂菲反应活性差,深度加氢过程难以进行。因此,设计高性能加氢饱和催化剂是整个反应的关键。
芳烃催化加氢反应过程中,H2活化是前提,反应物吸附、产物脱附是关键。菲加氢为逐环加氢,随饱和环数增加,末环加氢反应变难。芳烃分子在过渡金属上的吸附与炔烃、烯烃的吸附机理相似,符合d-π*反馈机制,芳烃化合物吸附强度与金属表面轨道杂化程度以及过渡金属d带的电荷密度相关,过渡金属d电子的性质会影响吸附质在金属表面的吸附进而影响催化活性[9-13]。1995年,HAMMER等[14]提出d带中心理论,将活性中心的价电子结构与催化剂的催化性能关联,该理论认为,d能带中心与吸附质在活性金属表面上的化学吸附相关。d带中心相对于费米能级位置越高,反键态占据的能量位置越高,反应分子吸附越稳定。对于d区元素而言,价带填充越多,吸附质与金属结合越弱,为获得最优活性,需在强吸附和快速解吸之间平衡。因此,d能带中心往往与吸附质的吸附能成典型的火山型曲线关系。近年来,研究者通过试验证实了d电荷密度可做为催化活性的描述符。基于d电荷密度描述符,LI等[15]利用吸附诱导表面重排法,通过控制Au、Pd含量合成了不同Au、Pd含量的AuxPd1-x双金属催化剂。掺杂Au显著改变了Pd电子结构,Pd质量分数为50%时,Pd的d电荷密度增益最大,为0.28 e。在喹啉选择性加氢反应中,Au50Pd50催化剂转换频率(fTO)值最高,负活化熵(ΔS0*)值最大,且AuPd纳米催化剂Pd d电荷密度与ΔS0*、fTO之间呈线性关系。SUN等[16]制备了C掺杂的Au负载在有序介孔碳上的C-Au/OMC催化剂,利用XANES(X-ray Absorption Near Edge Structure,X射线吸收近边结构)和XPS(X-ray Photoelectron Spectroscopy,X射线光电子能谱)技术表明C占据了晶格中空隙位置,促进了Au位点的电子转或重新分布,试验测得C-Au/OMC催化剂中Au的d电荷密度,在3-硝基苯乙烯选择性加氢反应中,将Au的d电荷密度与反应活性关联,得到d电荷密度与C-Au/OMC催化剂活性的关系。
研究表明,芳烃加氢反应中,催化剂活性金属的缺电子结构有利于菲加氢反应进行[17-23]。本课题组前期基于Ni基催化剂研究菲加氢反应,以TiO2掺杂Al2O3载体,通过次磷酸盐歧化法制备了Ni2P/TiO2-Al2O3催化剂,结果表明,掺杂TiO2降低了Ni2P催化剂Niδ+缺电子程度,提升了Ni2P/Al2O3催化剂菲加氢活性[24]。以Pt掺杂Ni,通过溶胶凝胶法制备了Pt-Ni/NiAlOx催化剂,结果表明,掺杂Pt使Ni/NiAlOx催化剂活性金属Ni处于缺电子结构,提升了催化剂菲加氢活性,但当Ni缺电子程度进一步增加时,加氢活性下降[25]。综上可知,催化剂电子结构是影响催化剂菲加氢性能的关键因素,但关于催化剂电子结构如何影响催化剂菲加氢性能的原因鲜有研究。
催化剂电子结构状态是催化加氢反应的重要影响因素,d电荷密度即d电子得失数量,反映d带空穴数量对吸附质在催化剂表面的吸附的影响。为此,笔者聚焦催化剂活性金属d电荷密度,基于Ni2P催化剂稳定性高、耐硫、耐氮性强等优势,采用次磷酸盐歧化法制备γ-Al2O3负载的Ni2P催化剂,以Ni2P/Al2O3催化剂为体系,通过调控P/Ni物质的量比,制备了晶相唯一(均为Ni2P晶相)、电子结构不同的Ni2P/Al2O3催化剂。探究了催化剂活性金属Ni d电荷密度如何影响反应分子吸附与催化剂菲加氢性能,明确Ni2P/Al2O3催化剂菲加氢反应合适的Ni d电荷密度。
1 试 验
1.1 Ni-xP/Al2O3催化剂的制备
采用次磷酸盐歧化法制备催化剂[26-27],依次加入次磷酸铵(NH4H2PO2)和乙酸镍(Ni(CH3COO)2·4H2O)的水溶液,全部溶解后,浸渍γ-Al2O3粉末,浸渍液中P/Ni物质的量比分别为1.5、1.8、2.0、2.5、3.0,基于前期研究结果[28],保持Ni含量为催化剂质量的10%,仅通过改变磷含量控制P/Ni物质的量比。将催化剂于室温中搅拌,直至浸渍液为胶状状态,随后转移至干燥箱在60 °C下干燥12 h,得到催化剂前驱体氧化物,在H2气氛下还原得到催化剂(H2流量:100 mL/.min,400 °C下恒温2 h)。不同P/Ni比Ni2P/Al2O3催化剂记为Ni-xP/Al2O3(x分别为1.5、1.8、2.0、2.5、3.0)。
1.2 Ni-xP/Al2O3催化剂的表征
使用Cu靶辐射源(λ=0.154 06 nm,U=40 kV,I=40 mA),在Rigaku Ultima IV衍射仪(XRD)上分析催化剂物相结构,XRD扫描角度(2θ)为10°~90°,扫描速率4(°)/min。使用美国FEI Talos F200X G2型高分辨透射电子显微镜分析催化剂微观形貌、颗粒尺寸。使用美国康塔公司的Quantachrome Autosorb-iQ分析催化剂比表面积和孔结构并采用BET法和BJH法计算催化剂比表面积和孔径分布。通过CO脉冲吸附分析催化剂活性位点的量。NH3-TPD分析在Autochem II 2920化学吸附仪上进行,分析催化剂的酸量和酸强度。通过X射线光电子能谱分析催化剂表面元素化学状态,仪器型号为岛津Kratos Analytical AXIS Supra,使用单色Al Kα为X射线源,在高真空条件下分析测试。通过软X射线吸收近边光谱(S-XANES)分析催化剂活性金属Ni d电荷密度。XANES谱在上海同步辐射光源(SSRF)SiP. ME2P站台02B02光束线上进行,在透射模式下记录Ni L边近边吸收谱。Ni-xP/Al2O3催化剂其他详细表征参见前期研究结果[26]。
1.3 催化剂活性评价
催化剂活性评价在连续型高压固定床反应器上进行,反应前对催化剂进行原位还原,在100 mL/min H2气流中以2 ℃/min升温速率加热至400 ℃,保持2 h,然后在连续H2气流中自然冷却至反应温度进行加氢反应。菲加氢反应操作条件具体如下:1%菲/十氢萘溶液,5 MPa,280~320 ℃,H2流量60 mL/min,原料进料量为6 mL/h,催化剂用量为4 mg,重时空速(WHSV)1 309/h。对称八氢菲加氢反应条件如下:1%对称八氢菲/十氢萘溶液,5 MPa, 280~320 ℃,H2流量60 mL/min,原料进料量6 mL/h,催化剂用量3 mg,重时空速1 746 h-1。每隔1 h反应产物取样并且分析。使用岛津GC-2010气相色谱分析加氢产物,选择正十四烷为内标物定量分析。
菲或对称八氢菲转化率计算公式如下:
(1)
式中,X为菲或对称八氢菲转化率,%;Nin为原料中菲或对称八氢菲的物质的量,mol;Nout为产物中菲或对称八氢菲的物质的量,mol。
菲或对称八氢菲加氢产物选择性计算公式如下:
(2)
式中,Sj为菲或对称八氢菲加氢产物选择性;Nj为菲或对称八氢菲某加氢产物的物质的量,mol;∑Nj为菲或对称八氢菲所有加氢产物物质的量的总和,mol。
菲加氢、对称八氢菲加氢反应fTO计算公式如下:
fTO=Fx/(n(Ni)f(Ni))=r0/f(Ni),
(3)
式中,F为反应原料摩尔流量,mol/s;x为全氢菲选择性,%;n(Ni)为催化剂中Ni的物质的量,mol;f(Ni)为活性金属Ni的分散度;r0为初始反应速率,催化反应的初始反应速率为单位时间单位摩尔活性金属转化的反应分子的量,s-1。
若反应为零级反应,则反应常数可近似为初始反应速率,即r0=k:
fTO=Fx/(n(Ni)f(Ni))=k/f(Ni)
(4)
1.4 反应动力学参数测定
对不同Ni-xP/Al2O3催化剂进行菲加氢反应、对称八氢菲加氢反应动力学分析,先确定加氢反应的反应级数,排除内外扩散(有关内外扩散排除的相关试验详见课题组前期研究[29]),改变反应原料浓度(原料质量分数分别为1%、2%、3%、4%的菲或对称八氢菲溶于十氢萘溶液中),进行加氢反应试验,计算初始反应速率,若反应速率不随反应原料浓度变化而改变,则反应近似为零级反应。然后计算不同温度下(280~320 ℃)的反应速率,绘制阿伦尼乌斯曲线,根据拟合曲线方程斜率和截距求得Ea(活化能)和A(样品LⅢ、LⅡ白线峰积分面积),基于过渡态理论,求得吸附活化熵(ΔS0*),计算方法见式(5)~(10)[15,30-31]:
fTO以Eyring方程的形式为
(5)
式中,T为温度,K;kB为玻尔兹曼常数;h为普朗克常数;R为普适气体常数;ΔH0*为焓变。
Ea可通过Temkin方程与ΔH0*相关联,即
Ea=ΔH0*+∑niΔHi,
(6)
式中,ni为反应级数;ΔHi为不同级数反应的焓变,当反应为零级反应时,Ea=ΔH0*。
则
(7)
由阿伦尼乌斯方程可得:
(8)
则
(9)
将式(4)、(9)关联,得活化熵变(ΔS0*)的计算公式为
(10)
1.5 Ni d电荷密度计算方法
Ni d电荷密度计算方法如下:利用XANES技术对催化剂的Ni L边进行表征,金属L边白线峰强度反映金属d带空穴量,与d带空穴量成正比。d带空穴量的变化由d带电荷密度变化引起。因此,以Ni/Al2O3催化剂为基准,通过对不同催化剂的XANES特征曲线进行积分计算得到d带空穴的变化量,进一步求得催化剂d电荷密度。d电荷密度记为d charge。催化剂活性金属Ni的d电荷密度具体计算方法见式(11)和(12)[32-34]:
(11)
(12)
式中,Δnholes为d带空穴的变化量;LⅢ(Ni)、LⅡ(Ni)为Ni的2个特征峰;A为样品LⅢ、LⅡ白线峰积分面积;σ/n3d为Ni 3d轨道每摩尔空位吸收截面,σ/n3d=13.73;σ(Ni)-σ(Cu)为Ni的LⅢ、LⅡ白线峰积分面积与Cu的LⅢ、LⅡ白线峰积分面积差减;n3d为3d带空穴数目;n3d(Ni),Ni 3d带空穴数目为1.18;n3d(Cu),Cu 3d带空穴数目为0.27。
2 结果与讨论
2.1 Ni-xP/Al2O3催化剂结构表征
2.1.1 Ni2P晶相的确定
通过XRD对不同P/Ni物质的量比(1.33、1.50、1.80、2.00、2.50、3.00)的Ni-xP/Al2O3催化剂进行定性分析,确定Ni-xP/Al2O3催化剂生成Ni2P晶相所需P/Ni比。研究表明,次磷酸盐歧化法在最佳还原温度(400 ℃)下制备的Ni2P/Al2O3催化剂XRD谱图中观察不到Ni2P衍射峰,因为Ni2P处于较高分散状态[28,35-38]。
为明确不同P/Ni比Ni-xP/Al2O3催化剂是否生成Ni2P晶相,提高催化剂还原温度,考察了550 ℃还原温度下制备的Ni-xP/Al2O3催化剂晶相结构。Ni-xP/Al2O3催化剂(550 ℃)还原XRD如图1所示,所有催化剂均出现明显Al2O3衍射峰,对比Ni2P(PDF#03-0953)标准卡片可知,除Ni-1.33P/Al2O3催化剂外,其余Ni-xP/Al2O3(x=1.5、1.8、2.0、2.5、3.0)催化剂在2θ为54.2°处显示出Ni2P特征衍射峰,且随着P/Ni比增大,Ni2P衍射峰强度变强。可以看出以次磷酸盐歧化法制备Ni2P/Al2O3催化剂,调控P/Ni比大于1.33,可形成Ni2P晶相。
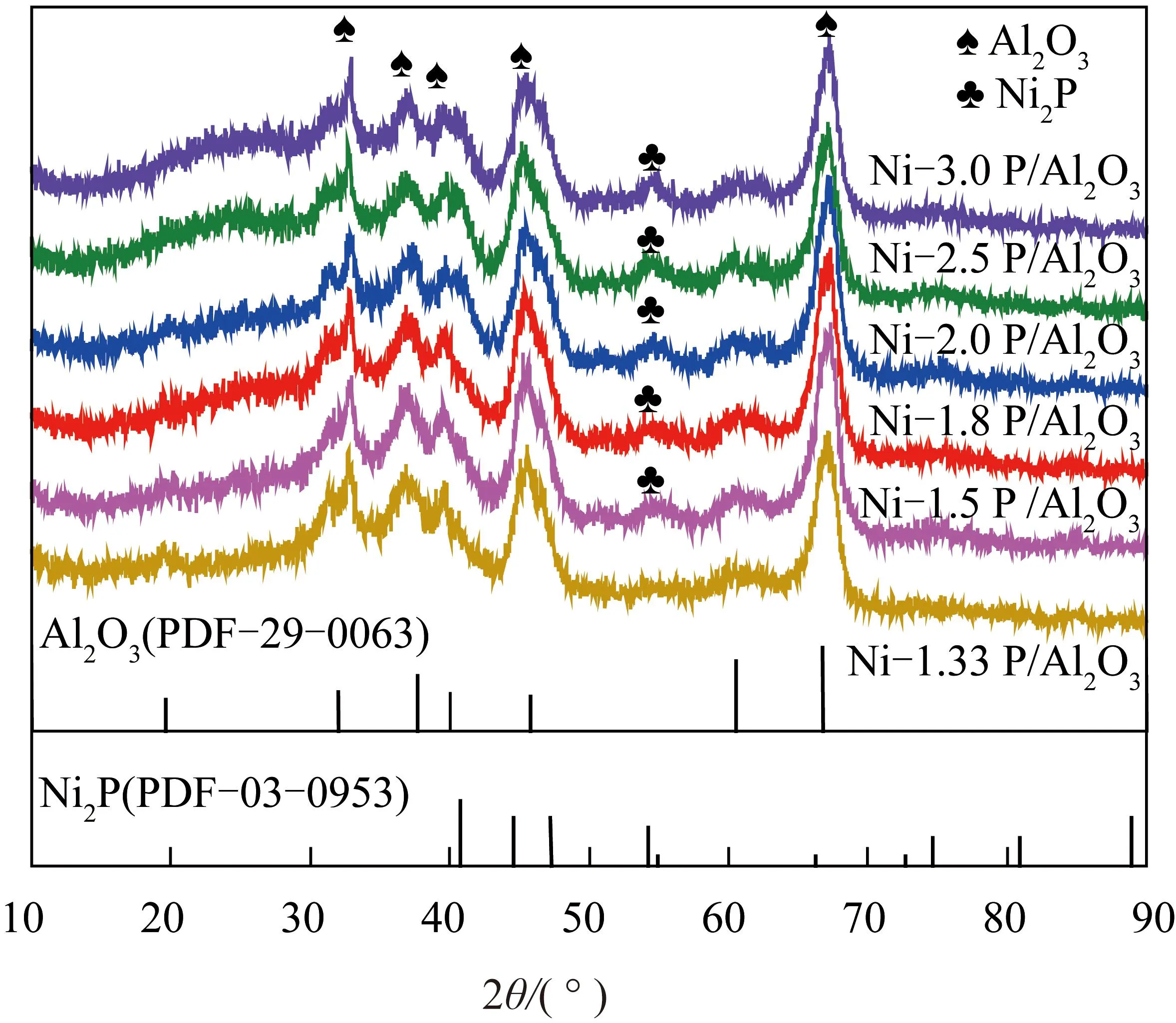
图1 Ni-xP/Al2O3催化剂(550 ℃)还原XRD图
2.1.2 表面元素电子性质分析
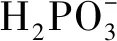

图2 Ni-xP/Al2O3催化剂XPS能谱图
分析不同P/Ni物质的量比Ni-xP/Al2O3催化剂Ni 2p结合能,具体见表1。由表1可以看出,Ni-xP/Al2O3催化剂Niδ+结合能随P/Ni物质的量比增加向高能处偏移,其中,Ni-1.5P/Al2O3催化剂Niδ+结合能最小为852.1 eV,Ni-3.0P/Al2O3催化剂Niδ+结合能最大为853.2 eV,表明随P/Ni物质的量比增大,Ni缺电子程度变大,催化剂电子结构发生变化。

比较不同P/Ni物质的量比Ni-xP/Al2O3催化剂XPS谱图,对各物种的峰面积积分,进行定量分析具体见表2。由表2可知,P/Ni物质的量比由1.5增至2.0时,Niδ+、Pδ-峰面积百分比随P/Ni物质的量比增大而增大,当P/Ni物质的量比大于2时,Niδ+、Pδ-峰面积百分比随P/Ni物质的量比进一步增大而变小,表明P/Ni物质的量比为2时,Ni-2.0P/Al2O3催化剂生成最多活性物种,有利于暴露出更多活性位点。

表2 Ni-xP/Al2O3催化剂Niδ+和Pδ-峰面积
为进一步探究不同P/Ni物质的量比对催化剂电子结构的影响,通过X射线吸收近边结构谱(XANES)技术表征Ni-xP/Al2O3催化剂Ni L边。Ni/Al2O3及Ni-xP/Al2O3催化剂Ni L边XANES谱图如图3所示,由P/Ni物质的量比变化引起的催化剂电子结构的差异可被观察,不同催化剂均在852.5 eV处出现一条明显的白线,白线峰的强度反映金属d带空穴数的变化,白线峰强度越强,d带空穴数越多。其中,Ni-1.5P/Al2O3催化剂白线峰强度最弱,表明该催化剂的Ni 3d带表现出较少空穴,随P/Ni物质的量比逐渐增加,催化剂白线峰强度增强,表明产生了更多空穴,其中,Ni-3.0P/Al2O3催化剂表现出最高的Ni 3d带空穴数。可知不同P/Ni物质的量比Ni-xP/Al2O3催化剂电子结构发生变化,且随P/Ni物质的量比增加,催化剂Ni d电荷密度变低,缺电子程度变大,与XPS表征结果一致。
图3 Ni/Al2O3及Ni-xP/Al2O3催化剂Ni L边XANES谱图
2.2 Ni-xP/Al2O3催化剂活性评价
2.2.1 Ni-xP/Al2O3催化剂菲加氢活性
排除内外扩散的前提下,在320 ℃、5 MPa、氢油比600、重时空速1 309 h-1,考察不同Ni-xP/Al2O3催化剂的菲加氢本征活性,反应物为1%菲/十氢萘溶液。菲加氢饱和反应路径[39]参考如图4所示。

图4 菲加氢反应网络路径[39]
Ni-xP/Al2O3催化剂的活性评价如图5所示。由图5(a)可知,Ni-2.5P/Al2O3催化剂菲加氢活性明显优于其他催化剂。以反应第5小时活性评价结果为对象,Ni-1.5P/Al2O3催化剂菲转化率为11.26%,全氢菲选择性为12.45%,随着P/Ni比增加,催化剂菲加氢活性提高,Ni-2.5P/Al2O3催化剂菲加氢转化率达到最高19.12%,全氢菲选择性为19.85%,但随P/Ni比进一步增加,催化剂菲加氢活性下降,Ni-3.0P/Al2O3催化剂菲加氢转化率降至17.05%,全氢菲选择性降至16.55%。考虑到不同P/Ni比催化剂上活性位点数量差异对Ni-xP/Al2O3催化剂菲加氢活性的影响,将活性归一化到单位活性中心Ni原子上,计算Ni-xP/Al2O3催化剂菲加氢反应fTO以比较催化剂本征活性。由图5(b)可知,Ni-1.5P/Al2O3、Ni-1.8P/Al2O3、Ni-2.0P/Al2O3、Ni-2.5P/Al2O3、Ni-3.0P/Al2O3催化剂fTO分别为20.98×10-3、23.07×10-3、26.63×10-3、44.64×10-3、35.65×10-3s-1,Ni-2.5P/Al2O3催化剂fTO最高,表明,Ni-2.5P/Al2O3催化剂在单位活性中心Ni原子上有最高菲加氢活性。因为具有适宜电荷密度的Ni可加强反应过程中菲与催化剂活性中心相互作用,促进菲的吸附,提升催化剂菲加氢活性。
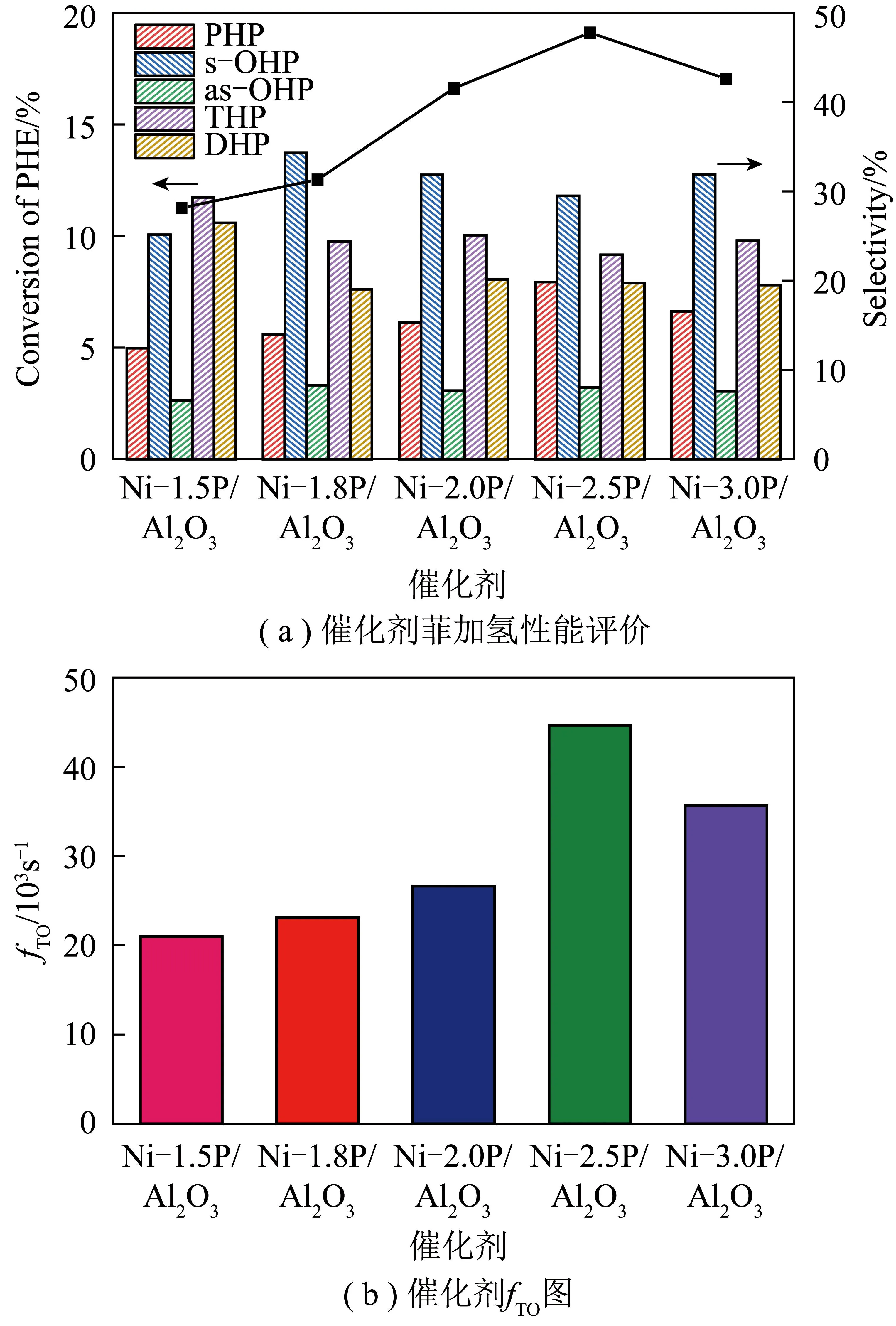
图5 Ni-xP/Al2O3催化剂的活性评价
进一步考察Ni-xP/Al2O3催化剂菲加氢转化率和全氢菲选择性随时间变化,探究不同Ni-xP/Al2O3催化剂稳定性。在320 ℃、5 MPa、重时空速1 309 h-1下,反应结果如图6所示,随着反应时间由1 h增至5 h,Ni-xP/Al2O3催化剂菲转化率及全氢菲变化范围均在初始活性10%左右,表明改变P/Ni物质的量比不会影响Ni-xP/Al2O3催化剂稳定性。
2.2.2 不同Ni-xP/Al2O3催化剂上菲的吸附活化熵
为探究催化剂活性金属Ni d电荷密度对菲加氢性能的影响,对菲加氢反应进行动力学分析。菲加氢反应是逐环加氢过程,全氢菲为目标产物,为保证菲加氢反应动力学的准确性,排除内扩散和外扩散的基础上,研究菲加氢反应级数,分别选取菲浓度0.05、0.10、0.15、0.20 mol/L,进行菲加氢反应,考察改变反应浓度对初始反应速率的影响(图7(a))。由图7(a)可知,随着菲浓度增加,初始反应速率几乎相同,不随反应物浓度变化而变化,菲加氢反应可近似看作零级反应。零级反应反应常数k可近似等于初始反应速率r0。

图7 菲加氢反应级数确定
吸附活化熵(ΔS0*)是过渡态的熵与反应物熵之间差值,由于催化剂表面与被吸附物种相互作用会导致自由度损失,因此活化熵可反映分子吸附到催化剂表面的活化程度,即吸附强度的变化[40]。对菲加氢反应进行动力学分析,改变反应温度280~320 ℃,计算不同温度下Ni-xP/Al2O3催化剂的反应速率常数,通过阿伦尼乌斯方程计算不同催化剂氢菲加氢反应Ea和指前因子A,求得ΔS0*,计算方法详见第1.4节。不同温度下不同催化剂的菲加氢活性评价如图7(b)所示,280 ℃时,Ni-xP/Al2O3催化剂菲加氢活性最低,随反应温度增加,菲转化率增加,全氢菲选择性增加,320 ℃时,催化剂的菲加氢活性最高,不同温度下,随着P/Ni比增加,催化剂菲转化率和全氢菲选择性先增加后减小,P/Ni比为2.5时,Ni-2.5P/Al2O3催化剂加氢性能最优。
计算不同温度下Ni-xP/Al2O3催化剂菲加氢反应速率常数,绘制阿伦尼乌斯曲线图(图8(a)),计算反应Ea、A。其中,Ni-1.5P/Al2O3催化剂菲加氢反应活化能最大,为34.59 kJ/mol,随着P/Ni物质的量比增加,催化剂菲加氢反应活化能呈倒火山型曲线(图8(b)),当P/Ni物质的量比为2.5时,Ni-2.5P/Al2O3催化剂菲加氢反应活化能最小,为23.52 kJ/mol,有利于进行菲加氢反应。吸附活化熵(ΔS0*)的正负代表反应体系中自由度增加或损失,催化反应中反应分子在催化剂表面吸附过程会导致自由度损失,自由度损失越多,活化熵绝对值越大,吸附越强。菲加氢反应中,不同催化剂负活化熵随P/Ni物质的量比增加呈火山型曲线,Ni-2.5P/Al2O3催化剂有最大负活化熵,-223.23 J/(mol·K),表明菲在Ni-2.5P/Al2O3催化剂上吸附最强(图8(b))。反应分子吸附强度与活性金属d轨道电荷密度相关,活化熵反映反应分子在催化剂表面的活化程度,也受活性金属d电荷密度的影响。此外,还可发现活化熵值随活化能减小而减小,二者存在良好线性关系,称为补偿效应。因为在吸附过程中系统自由度变化,随着系统能量减小,活性金属原子与反应分子之间相互作用变强[41]。通过调控金属d电荷密度调变反应分子菲的吸附,影响催化剂菲加氢活性,Ni-2.5P/Al2O3催化剂有最小活化能,最大的负活化熵和最高的fTO,这表明菲加氢反应中,Ni-2.5P/Al2O3催化剂可活化更多菲分子,且对菲的吸附最强,促进了菲的转化,表现出最高的菲加氢活性。
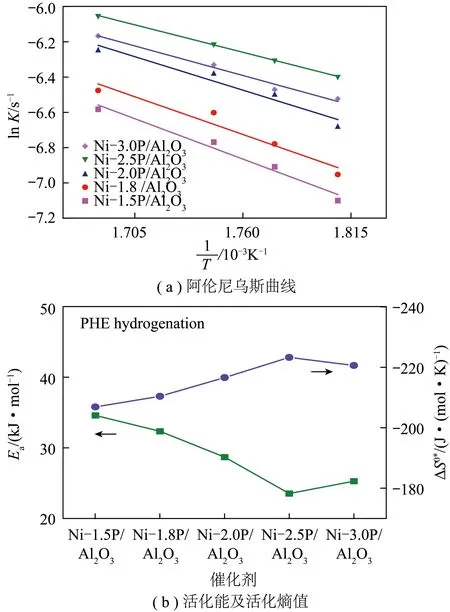
图8 Ni-xP/Al2O3催化剂上菲加氢反应速率常数
2.2.3 Ni-xP/Al2O3催化剂对称八氢菲加氢反应活性
菲加氢反应过程中,对称八氢菲到全氢菲的加氢是整个加氢反应的速控步骤,为探究不同P/Ni摩尔比对Ni-xP/Al2O3催化剂对称八氢菲加氢活性的影响,考察了系列Ni-xP/Al2O3催化剂对称八氢菲加氢反应本征活性,在320 ℃、5 MPa、氢油比600、重时空速1 746 h-1下,反应物为1%对称八氢菲/十氢萘溶液。催化剂对称八氢菲加氢性能评价如图9(a)所示,反应5 h后,Ni-1.5P/Al2O3催化剂对称八氢菲加氢性能最优,对称八氢菲转化率14.72%,全氢菲选择性44.69%。随着P/Ni比增加,Ni-xP/Al2O3催化剂的对称八氢菲加氢活性降低,P/Ni比为2.5时,Ni-2.5P/Al2O3催化剂对称八氢菲加氢活性最低,对称八氢菲转化率9.93%,全氢菲选择性31.27%。

图9 Ni-xP/Al2O3催化剂的活性评价
进一步比较Ni-xP/Al2O3催化剂对称八氢菲加氢本征活性,将活性归一化到单位活性中心Ni原子上,计算fTO。催化剂fTO如图9(b)所示,Ni-1.5P/Al2O3催化剂fTO最高,117.09×10-3s-1,不同催化剂对称八氢菲加氢活性排序Ni-1.5P/Al2O3>Ni-1.8P/Al2O3>Ni-2.0P/Al2O3>Ni-3.0P/Al2O3>Ni-2.5P/Al2O3,与催化剂菲加氢活性呈相反趋势。由于菲与对称八氢菲自身π电子密度的差异,导致菲、对称八氢菲在催化剂表面吸附时对活性金属电荷密度需求不同,表现为菲、对称八氢菲在Ni d电荷密度不同的Ni-xP/Al2O3催化剂上吸附强度不同,表现出不同加氢活性。
2.2.4 不同催化剂上对称八氢菲的吸附活化熵
为进一步探究活性金属Ni d电荷密度对Ni-xP/Al2O3催化剂对称八氢菲加氢活性的影响,明确对称八氢菲在不同P/Ni物质的量比Ni-xP/Al2O3催化剂上吸附强度的差异,对不同催化剂对称八氢菲加氢反应进行动力学分析,改变反应温度280~320 ℃,计算吸附活化熵(ΔS0*)。图10为不同催化剂不同温度下对称八氢菲加氢活性评价。随温度增加,催化剂对称八氢菲加氢活性下降,低温有利于对称八氢菲加氢反应进行。在280 ℃,催化剂对称八氢菲加氢活性最高,全氢菲选择性最高。不同温度下,Ni-1.5P/Al2O3催化剂均有最优异的对称八氢菲加氢活性,280 ℃下,Ni-1.5P/Al2O3催化剂全氢菲选择性达到72%左右。随着P/Ni比增加,催化剂对称八氢菲加氢活性先减小后增加,Ni-2.5P/Al2O3催化剂对称八氢菲加氢活性最低,但也达到54%左右。

图10 不同温度下Ni-xP/Al2O3催化剂对称八氢菲加氢性能评价
不同催化剂对称八氢菲加氢反应阿伦尼乌斯曲线如图11(a)所示。其中,Ni-1.5P/Al2O3催化剂对称八氢菲加氢反应活化能最小,为36.36 kJ/mol,Ni-xP/Al2O3催化剂对称八氢菲加氢反应活化能随P/Ni物质的量比的增加呈火山型曲线(图11(b)),当P/Ni物质的量比为2.5时,Ni-2.5P/Al2O3催化剂活化能最大,为44.73 kJ/mol。对称八氢菲加氢反应中,不同催化剂负活化熵随P/Ni物质的量比增加呈倒火山型曲线,Ni-1.5P/Al2O3催化剂有最大负活化熵-199.76 J/(mol·K),说明对称八氢菲在Ni-1.5P/Al2O3催化剂上吸附最强。Ni-1.5P/Al2O3催化剂活化能最小,负活化熵最大,fTO最高,表明对称八氢菲加氢反应中Ni-1.5P/Al2O3催化剂可活化更多对称八氢菲分子,且对对称八氢菲吸附最强,因此对称八氢菲加氢活性最优。相反,Ni-2.5P/Al2O3催化剂活化能最大,负活化熵最小,对称八氢菲加氢活性最差。
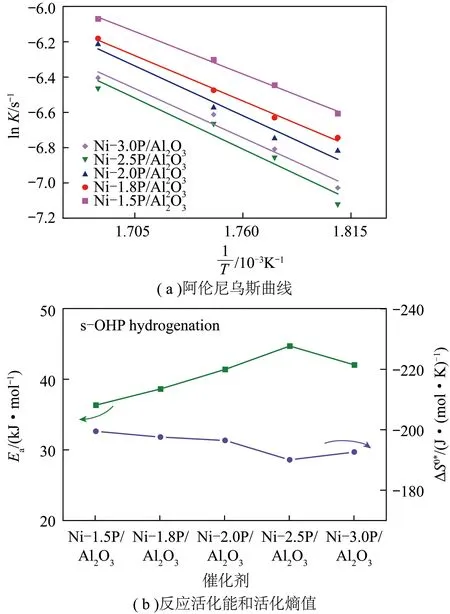
图11 Ni-xP/Al2O3催化剂上对称八氢菲加氢反应速率常数
2.2.5 活性金属Ni d电荷密度的定量计算
上述研究发现活性金属d电荷密度主要通过影响反应分子吸附来影响催化活性,且菲、对称八氢菲在不同Ni d电荷密度的Ni-xP/Al2O3催化剂上吸附强度不同。其中,菲加氢反应中,Ni-2.5P/Al2O3催化剂对菲的吸附最强,菲加氢活性最优;对称八氢菲加氢反应中,Ni-1.5P/Al2O3催化剂对对称八氢菲吸附最强,对称八氢菲加氢活性最优。结合催化剂加氢活性、反应分子吸附性能的变化,为进一步明确Ni-xP/Al2O3催化剂菲加氢、对称八氢菲加氢反应合适的Ni d电荷密度,对不同催化剂Ni L边XANES特征曲线积分量化d带空穴数,以Ni/Al2O3催化剂为基准,计算不同催化剂Ni d电荷密度(计算方法详见第1.5节),结果见表3。

表3 定量计算Ni-xP/Al2O3催化剂Ni d电荷密度
将Ni d电荷密度与催化剂加氢活性关联,图12(a)显示了不同催化剂Ni d电荷密度定量结果与其菲加氢活性之间关系,不同催化剂菲加氢反应fTO随Ni d电荷密度降低呈火山型趋势。催化剂Ni d电荷密度轻微降低表现为fTO的提升,但当Ni d电荷密度进一步降低时,催化剂菲加氢活性降低,菲加氢活性在Ni d电荷密度-0.24 e时最高。

图12 Ni d电荷密度与不同催化剂加氢性能关系
由图12(b)可知,不同催化剂对称八氢菲加氢反应fTO随Ni d电荷密度的降低呈倒火山型曲线,Ni d电荷密度为-0.05 e时,催化剂对称八氢菲加氢活性最高;Ni d电荷密度为-0.24 e时,催化剂对称八氢菲加氢活性最低。计算结果表明,由于菲、对称八氢菲自身结构差异,强化吸附时对d电荷密度需求不同,Ni-xP/Al2O3催化剂进行菲加氢反应时的适宜Ni d电荷密度为-0.24 e左右,进行对称八氢菲加氢反应适宜的Ni d电荷密度在-0.05 e左右。
3 结 论
1)采用次磷酸盐歧化法,调变前驱体P/Ni物质的量比,实现了Ni d电荷密度的调控(-0.05~-0.31 e),P/Ni物质的量比为1.5~3.0时,合成了晶相唯一(均为Ni2P)、不同电子结构的Ni2P/Al2O3催化剂。
2)320 ℃、5 MPa、空速1 309 h-1下,菲加氢反应活性评价显示,Ni-2.5P/Al2O3催化剂表现出菲加氢活性最优,转换频率fTO最高,44.64×10-3s-1。结合动力学分析及吸附活化熵计算,菲加氢反应中,Ni-2.5P/Al2O3催化剂活化能最小,负活化熵最大。可见,Ni-2.5P/Al2O3催化剂可活化更多菲分子,且对菲的吸附最强,促进了菲转化,菲加氢活性最优。
3)活性金属Ni d电荷密度主要通过影响菲的吸附活化影响加氢活性,通过调变Ni d电荷密度调变了菲在催化剂表面的吸附性能,菲吸附强度随Ni d电荷密度降低呈火山型曲线,适宜Ni d电荷密度有利于菲的吸附活化。
4)定量计算Ni d电荷密度发现,由于原料结构差异,强化菲、对称八氢菲吸附时对活性金属d电荷密度需求不同,Ni-xP/Al2O3催化剂进行菲加氢反应的适宜Ni d电荷密度约-0.24 e,进行对称八氢菲加氢反应适宜的Ni d电荷密度约-0.05 e。
猜你喜欢
睿士(2023年10期)2023-11-06
材料与冶金学报(2022年2期)2022-08-10
中学生数理化·中考版(2021年10期)2021-11-22
小学科学(学生版)(2021年3期)2021-04-13
小哥白尼(趣味科学)(2020年9期)2021-01-18
云南化工(2020年11期)2021-01-14
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
作文成功之路·小学版(2020年6期)2020-07-27
Coco薇(2015年5期)2016-03-29
新高考·高一物理(2015年6期)2015-09-28
