
以CeO2为载体的Fe基载氧体与CO反应机理模拟
2024-03-21 04:53陈建标霍兆义
洁净煤技术 2024年2期
穆 林,孙 萌,张 彬,尚 妍,东 明,陈建标,霍兆义
(1.大连理工大学 能源与动力学院 海洋能源利用与节能教育部重点实验室,辽宁 大连 116024;2.南京工业大学 能源科学与工程学院,江苏 南京 211816;3.辽宁科技大学 材料与冶金学院,辽宁 鞍山 114051)
0 引 言
化学链燃烧技术(Chemical Looping Combustion,CLC)是一种新型的近零碳排放燃烧技术,能够高效利用能量的同时实现低成本的CO2分离和捕集[1]。化学链燃烧系统由燃料反应器和空气反应器2部分组成,载氧体作为燃料反应器和空气反应器间的传递媒介,在2个反应器之间循环往复,同时进行晶格氧和热量的传递,反应产物CO2和H2O只需简单冷凝便可高效分离并捕集CO2。载氧体的本质特性是影响化学链燃烧系统运行效果的主要因素之一[2]。Fe基载氧体因其成本低廉、储量丰富、环保、机械性能好等优点受到广泛关注,被认为是最具发展潜力的载氧体之一;然而Fe基载氧体的低反应活性也制约其大规模应用[3-4],通过引入活性催化组分,调谐微观结构提高Fe基载氧体的反应性能是目前化学链领域的研究热点之一[5-6]。
CeO2(氧化铈)作为稀土元素氧化物,具有较高的催化反应活性[7],已广泛应用于固体氧化物燃料领域。CeO2具备良好的吸氧-释氧能力,会随氧化还原环境改变发生Ce4+和Ce3+的可逆转变,因此也被用作改良载氧体反应性能的材料[8-9]。此外,考虑到其在地壳中含量丰富,其价格优势推动铈化合物的利用。LIANG等[10]研究了掺入CeO2的Fe2O3/LaNiO3载氧体的反应稳定性和CH4转化率,结果表明,Fe2O3-CeO2/LaNiO3载氧体历经100次循环仍能保持较高活性;反应生成的CeFeO3能增加载氧体表面的晶格氧量。韩丹华等[11]开展了基于NiO-CeO2/γ-Al2O3复合载氧体的化学链重整制氢试验,结果表明,添加CeO2可增加载氧体的氧空位,削弱NiO与γ-Al2O3间的相互作用,扩大活性物质的分散。WANG等[12]研究表明,添加CeO2能促进CaSO4钙基载氧体中氧转移和交换,增强CaSO4载氧体的反应活性。范浩熙等[13]通过氧化还原循环试验分析了CeO2/NiO复合载氧体的反应性能,结果表明850 ℃焙烧下的CeO2/NiO载氧体具有良好的结构稳定性、优异的储放氧性能及较强的循环稳定性。
在理论计算与分析方面,基于密度泛函理论(Density Functional Theory,DFT)的理论研究可从分子或原子层面揭示气体分子在不同晶体结构的载氧体表面吸附、氧化、催化分解的作用机制,从而有助于阐明分子或原子级反应过程中的电子迁跃及分子结构演变特征[14]。梁志永等[15]采用基于DFT理论的CASTEP(Cambridge Serial Total Energy Package)模块搭建了添加Co的Fe基载氧体模型,并根据计算得到的表面能、掺混结合能、态密度等电子性质研究了Co的引入对Fe基载氧体结构性质和反应性能的影响,结果表明Co能促进Fe基载氧体典型掺混点位周围O原子的键长伸长,使其更易与CO分子结合生成CO2,从而降低反应能垒。FENG等[16]通过DFT计算了Li、Na、K、Rb和Cs掺杂剂等对Fe2O3晶体结构的调谐影响特性。结果表明,所研究掺杂剂均能均匀分布于Fe2O3载氧体表面,其中,Li、Na和K可显著增强Fe2O3表面氧的活性和反应性,而Rb和Cs的活性增强效果较弱。袁妮妮等[17]基于密度泛函理论分析了低浓度Cu的引入对Fe2O3载氧体与H2分子反应过程的影响。结果表明,Cu的掺混引起Fe2O3载氧体的结构改变,晶格氧释放能力提升;降低了Fe2O3载氧体与H2反应所需活化能,提高了Fe2O3反应活性。
针对CeO2作为基体材料,CHU等[18]采用理论分析了4种CeO2(111)表面的CO直接氧化过程。结果发现排列可增强阶跃边缘的CO吸附C—O键和表面Ce—O键,此外Ce 4f轨道能级的异常降低能促进CO氧化。ZHANG等[19]基于密度泛函理论计算了甲醛在Co调谐后的CeO2(111)表面的反应情况,从而揭示了甲醛的吸附氧化机理,获得了Co调谐增强甲醛在CeO2(111)的吸附特性及甲烷的最小动力学反应路径。氧载体反应性受到晶格氧从体到表面扩散的影响,相关研究表明,基于CeO2的高氧传导和高温烧结抗性,使其被认为是纯铁氧化物的合适载体[20]。
然而,CeO2作为活性催化载体对Fe基载氧体进行改性的理论研究较少,且以铁基载氧体研究CeO2的掺杂特性无法完整体现CeO2和Fe2O3的催化特性[21]。笔者以密度泛函理论为基础,以CeO2为活性催化载体,对Fe基载氧体进行催化调谐,从分子层面研究Fe基载氧体化学链燃烧过程中,纯净的Fe2O3及以CeO2为载体的Fe2O3复合载氧体的微观结构演变和内部电子迁移的规律,获得CeO2调谐Fe基载氧体及其与CO反应性能的内在反应机理和反应路径,为Fe基载氧体的设计制备和优化调谐提供理论指导,从而进一步提高Fe基载氧体反应活性和稳定性。
1 计算模型及方法
1.1 计算模型
基于密度泛函理论的Materials Studio 2018软件包含可视化模块和CASTEP模块,能分别实现晶体结构模型搭建和多种电子结构性质的计算。CASTEP模块的优势体现在采用平面波赝势法计算周期性模型获得的结果更加准确。广义梯度近似GGA(Generalized Gradient Approximation)下的PBE(Perdew-Burke-Ernzerhof)泛函能准确分析电子间复杂的相互作用[22]。设置的几何结构优化收敛标准参数如下:平面波截断能设置为380 eV,赝势为Ultrasoft。采用Monkhorst-Pack方法对第一布里渊区的高对称特殊k点进行积分,k点设置为2×2×1。能量、原子间最大相互作用力、原子所受最大应力和原子位移最大分别设置为2.0×10-5eV/atom、0.5 eV/nm、0.1 GPa及1×10-4nm。过渡态搜索采用线性同步转变LST(Linear Synchronous Transit)与二次同步转变QST(Quadratic Synchronous Transit)方法[23-24],过渡态判据为除固定原子外,相关原子的振动只有唯一虚频。
1.2 计算方法
反应系统的吸附能Eads计算公式为
Eads=EAB-EA-EB,
(1)
式中,EA为吸附前A体系能量,eV;EB为吸附前B体系的能量,eV;EAB为A与B吸附后的整体能量,eV。
活化能ΔEa计算公式为
ΔEa=ETS-EIS。
(2)
其中,ETS为反应过程中过渡态能量,eV;EIS为初始态的能量,eV。一般来说,活化能是指分子从常态转变为易发生化学反应的活跃状态所需能量,是衡量化学反应发生所需最小能量的指标,活化能越小,反应越容易发生。
反应热ΔE是指在反应过程中,系统与外界环境交换的能量,计算公式为
ΔE=EFS-EIS。
(3)
其中,EFS为反应终态的能量,eV。系统最终状态能量小于初始状态能量时,反应放热,ΔE<0;否则,反应为吸热反应,ΔE>0。
1.3 模型建立
在组合物模型搭建过程中,以活性组分CeO2作为模型载体,将载氧体Fe2O3负载到载体CeO2表面,用以提高模型构建的可靠性和计算效率[25-26]。经结构优化后的Fe2O3团簇为圆锥形结构(图1(a)),其晶体结构对称性好,结构稳定。CeO2为面心立方的萤石结构,晶胞由4个Ce原子和8个O原子组成,其晶胞结构属于Fm-3m空间群,晶胞参数α=β=γ=90°,棱长a=b=c=0.548 nm,具体如图1(b)所示。

图1 Fe2O3/CeO2组合物模型的搭建
由于CeO2(111)晶面热力学性质最稳定[26],与自然界存在的晶面理化性质最接近,因此选择CeO2原胞的111表面进行切割,并在该表面基础上进行2×2超胞,建立4层周期性平板模型,底部2层固定,上面2层进行弛豫。为消除层与层间的相互影响,真空层厚度设置为1.5 nm,CeO2(111)表面模型如图1(c)所示;Fe2O3团簇负载到CeO2(111)表面,形成Fe2O3/CeO2组合物模型,如图1(d)所示。LI等[27]基于XRD(X-ray Diffraction)和H2-TPR(Temperature Programmed Reduction)对共沉淀法制备的Fe2O3/CeO2复合载氧体进行分析,结果表明Fe2O3/CeO2复合氧化物晶格氧含量较高,反应活性和选择性良好。
CeO2晶胞几何优化前后的参数见表1,可知GGA-PBE泛函对CeO2晶格参数计算的相对误差均小于4%,表明泛函、收敛值和k点等参数的设定较合理,同时对基于CeO2晶胞结构进行的多种结构优化提供合理性依据。CeO2晶胞结构优化后的参数略高于试验参数,这是由于PBE泛函会使键长和晶格常数偏大。

表1 CeO2结构优化前后参数
2 结果与讨论
2.1 CO在Fe2O3团簇表面吸附的机理
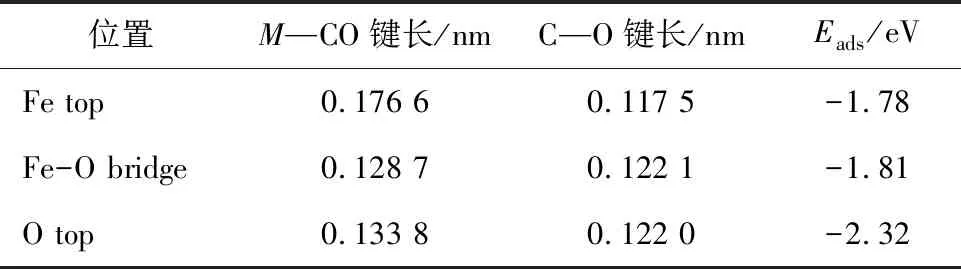
Fe2O3团簇表面电子分布不均匀,不同吸附位所表现的性质存在差异。为研究CO在Fe2O3团簇表面不同吸附位的吸附差异性,需选择Fe2O3团簇表面典型的吸附位作为研究对象。Fe2O3团簇表面分布3种典型的吸附位置,如图2(a)所示。位于Fe2O3团簇中Fe原子和O原子上的吸附位置分别命名为Fe顶位(Fe top)和O顶位(O top),Fe原子和O原子间的吸附位置命名为Fe—O桥位(Fe—O bridge)。CO在Fe2O3团簇表面Fe top、Fe—O bridge和O top吸附位的吸附构型如图2(b)~2(d)所示,CO在Fe2O3团簇3个吸附位的几何参数见表2。由表2可知,CO在Fe2O3团簇Fe top的吸附距离最长,吸附能最小且低于1 eV,属于典型的物理吸附[28];与Fe top吸附位置相比,CO在Fe2O3团簇Fe—O位的吸附距离减小了0.065 nm,吸附能增加了1.2 eV,较弱的物理吸附转变为化学吸附,CO吸附效果改善;CO在Fe2O3团簇O top处的吸附距离进一步减小,吸附能增至-2.69 eV,吸附结构更加稳定,进一步提高对游离CO分子的吸附效果。

表2 CO在纯净Fe2O3团簇表面吸附时的几何参数

图2 CO在Fe2O3团簇表面3个吸附位的吸附
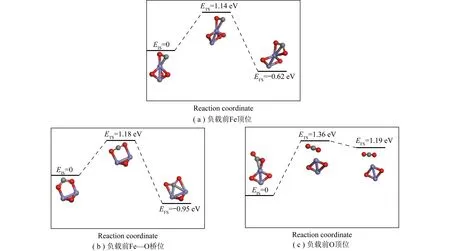
从C—O键伸长看,CO在自由状态下的C—O键长为0.115 7 nm,CO在Fe2O3团簇Fe等位、Fe—O桥位和O顶位处的键长均有不同程度伸长,尤其是CO在Fe—O桥位和O顶位处键长伸长程度明显大于Fe顶位。由吸附能可知,Fe2O3团簇表面Fe顶位、Fe—O桥位和O顶位3个吸附位对CO吸附的效果为:Fe顶位 CO在纯Fe2O3团簇表面反应路径的起点为Fe2O3团簇表面的CO吸附,对应的能量定义为0;反应路径的终点为CO与Fe2O3团簇反应后生成CO2和Fe2O2团簇;反应过程中需吸收能量用于克服旧键断裂,能量吸收的最高点物质定义为过渡态,对应的能量为活化能。CO在Fe2O3团簇不同吸附位置的反应路径如图3所示。 图3 CO与Fe2O3团簇不同位置的反应路径 从反应的活化能看,越靠近O端,反应活化能越高。经计算,CO在Fe2O3团簇表面Fe顶位、Fe—O桥位和O顶位反应的活化能分别为1.14、1.18和1.36 eV;CO在Fe2O3团簇表面Fe顶位和Fe—O桥位反应释放的能量分别为0.62、0.95 eV,而CO在Fe2O3团簇表面O顶位吸收的能量为1.19 eV,这与CO在Fe2O3团簇表面的吸附位置及吸附构型有关。较大的吸附能有助于Fe2O3团簇捕捉到更多游离的CO分子并与之结合,但吸附能越大也意味着吸附结构越稳定,使得后续反应过程中打破原有化学键的难度更大,需要吸收更多能量,因此会增大反应的活化能。根据表2吸附能Eads可知,Fe顶位、Fe—O桥位和O顶位的活化能随吸附能增加而增加,二者呈正相关。反应过程中新键形成所释放的能量一部分用于弥补活化能,一部分作为反应热。新键形成的释放热大于活化能时,反应放热;小于活化能时,反应吸热。因此,CO在Fe2O3团簇表面Fe顶位、Fe—O桥位反应释放能量,而在O顶位反应吸收能量[24]。 Fe2O3团簇负载到CeO2(111)表面后的微观结构如图4(a)所示,差分电荷密度如图4(b)所示,黄色区域表示失去电子,蓝色区域表示获得电子。可知Fe2O3团簇中的Fe原子位于失去电子的黄色区域,而Fe2O3团簇与CeO2(111)表面之间形成化学键的区域主要为蓝色区域,表现为获得电子。表明Fe2O3团簇负载到CeO2(111)表面后,Fe2O3团簇中Fe原子的电子向CeO2(111)表面迁移,用于二者形成化学键。 图4 Fe2O3团簇负载CeO2(111)表面的微观结构及电子性质 经计算,Fe2O3团簇与CeO2(111)表面吸附能较高,为-3.92 eV,二者较高的化学键强度表明Fe2O3团簇与CeO2(111)表面通过化学吸附形成了紧密结合体。Fe2O3团簇的平均键长由负载前的0.180 1 nm伸长到0.183 3 nm,负载后Fe2O3团簇整体化学键强度减弱,反应活性提高。顾振华等[29]采用共沉淀法制备CeO2与Fe2O3复合载氧体,并研究了复合载氧体在甲烷化学链燃烧中的反应稳定性和反应活性,研究发现CeO2能与Fe2O3充分混合并产生较大的接触界面,促使Fe2O3中内部深层晶格氧释放并向表面迁移,将Fe2O3还原为价态更低的铁氧化物,这与本文理论计算结果吻合。 Fe2O3团簇及其负载到CeO2(111)表面后的p轨道和d轨道如图4(c)、4(d)所示。负载后d轨道的DOS图发生平移,Fe2O3团簇负载到CeO2(111)表面后,其p和d轨道的态密度曲线变得平缓,经计算可知,纯净Fe2O3团簇p轨道的DOS面积为29.7(单位为电子数量),而负载到CeO2(111)表面后的Fe2O3团簇p轨道的DOS积分面积减小至20.1,d轨道的DOS积分面积由20.4降至19.5,所包围的面积减小表明Fe2O3团簇p和d轨道均体现出失去电子的特征。Fe2O3团簇p轨道和d轨道的态密度曲线呈现一定程度向费米能级方向偏移的趋势,以图4(d)为例,基于d带中心理论[30]可知,其中心位置由未负载时的-1.21 eV移到负载后的-1.18 eV,表明负载后吸附强度有所增强。Fe2O3团簇d轨道中现存的电子向远离电子核的方向迁移,受到的原子核束缚作用降低,使负载后的Fe2O3团簇d轨道的电子更易发生迁移,化学活性更好[31]。 Fe2O3团簇负载CeO2(111)表面后CO吸附的几何参数见表3。结合表2、3可知,CO在Fe2O3团簇Fe—O桥位和Fe顶位2个吸附位的吸附能有所提升,吸附更稳定,而O顶位的吸附能减小。这是由于Fe2O3团簇Fe顶位失去电子较多,带正电更多,从而表现为对CO的吸附作用加强;而Fe2O3团簇O顶位原子得到电子,带负电更多,从而表现为对CO的吸附作用减弱。 表3 Fe2O3团簇负载CeO2 (111)表面后CO吸附的几何参数 Fe2O3团簇负载CeO2(111)表面后CO的吸附结构及电荷差分密度如图5所示。可知Fe2O3团簇与CO分子之间形成的化学键被蓝色区域包裹,获得大量电子;而CO分子中C—O键主要处于黄色区域中,表现为失去电子,强度减弱,这是CO吸附在Fe2O3团簇后自身键长相比自由状态伸长的主要原因。 图5 Fe2O3团簇负载CeO2(111)表面后CO的吸附结构及差分电荷密度 CO与Fe2O3/CeO2组合物模型在3个吸附位的反应路径如图6所示。由图6(a)可知,在Fe2O3团簇负载CeO2(111)表面后,Fe顶位处反应的活化能降低0.04 eV,释放的能量减少了0.53 eV,主要原因是负载后Fe2O3团簇成键强度减弱,表面O原子更易脱离,并向CO方向迁移形成新键,因此活化能降低;另一方面,负载后的Fe2O3团簇对CO的吸附作用更强,CO从Fe2O3团簇表面断键脱附需要更多能量,而新键形成释放的能量多用于弥补脱附所需能量,使释放能量降低。由图6(b)可知,Fe—O桥位处反应活化能降低0.71 eV,释放的能量减少1.05 eV,主要原因是CO在Fe2O3团簇表面以碳酸盐形式吸附[32],相关原子之间的迁移距离减少,因此相关化学键断裂所需能量减少,活化能明显下降。但后期形成的CO2脱附离开表面,还需能量克服Fe2O3团簇表面的束缚,释放的能量降低。由图6(c)可知,O顶位处反应的活化能降低了0.80 eV,吸收的能量减少了0.92 eV,这是因为Fe2O3团簇活化,断键更容易;同时负载后,Fe2O3团簇对CO的吸附作用减弱,使得CO从Fe2O3团簇表面脱附所需能量更少,二者的综合作用表现为反应活化能降低和释放能量增加。MA等[33]采用不同含量CeO2修饰赤铁矿,并使用流化床试验台测定修饰后赤铁矿的氧化还原性和反应稳定性。研究发现CeO2促进赤铁矿中晶格氧的迁移能力,提高了赤铁矿的反应活性。ZHU等[34]制备复合载氧体用于化学链循环反应生产合成气。研究发现CeO2使Fe2O3载氧体历经多次氧化还原循环反应仍能保持良好的反应活性和反应稳定性。 图6 CO与Fe2O3/ CeO2组合物在3个吸附位的反应路径 从活化能角度,对于纯Fe2O3团簇而言,CO较易在Fe顶位反应,且反应迅速;在Fe2O3/CeO2组合物条件下,CO反而不易在Fe顶位反应,活化能明显高于其他吸附位。从结构分析,Fe2O3/CeO2组合物使CO吸附位置更倾向于从Fe2O3侧面进攻,提高接触面积。总体来说,CeO2的引入能明显降低3个吸附位所需活化能,一定程度上促进反应进行[27]。 1)Fe2O3团簇稳定结合在CeO2(111)表面。Fe2O3团簇电子向CeO2(111)表面转移,并分布于二者表面间的化学键周围,从而形成较强的化学键。 2)Fe2O3团簇负载CeO2(111)表面后,CO在Fe2O3团簇表面Fe—O桥位和Fe顶位2处吸附效果改善,吸附能增大;CO在Fe2O3团簇表面O顶位过强的吸附作用减弱。 3)CO分子在Fe2O3/CeO2复合载氧体的Fe2O3团簇3个吸附位反应的活化能均降低,原因是CeO2活化Fe2O3团簇的键长,使Fe2O3团簇断键难度降低,破坏旧键所吸收的能量更少;CO分子在Fe2O3团簇表面以碳酸盐形式吸附,使团簇表面原子迁移距离减小,并与CO分子形成新的化学键,因此能量克服迁移的势能阻力降低;CO分子在Fe2O3团簇表面过强的吸附作用被削弱,有利于后期CO2分子脱离Fe2O3团簇表面。2.2 CO在纯Fe2O3团簇表面的反应路径
2.3 Fe2O3团簇负载CeO2(111)表面后的微观结构特性

2.4 CO的吸附及电荷差分密度分析

2.5 CO在Fe2O3/CeO2组合物模型表面的反应路径

3 结 论
猜你喜欢
华北电力大学学报(自然科学版)(2022年4期)2022-08-17
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
燃烧科学与技术(2021年5期)2021-10-28
山东理工大学学报(自然科学版)(2021年6期)2021-07-02
洁净煤技术(2021年2期)2021-04-08
西部交通科技(2020年3期)2020-06-19
西南交通大学学报(2016年4期)2016-06-15
公路与汽运(2016年3期)2016-06-08
