
性腺母细胞瘤的分子遗传机制研究进展
2015-10-22 05:09余莉莉董琬如陈明会孔祥阳
遗传 2015年11期
余莉莉,董琬如,2,陈明会,孔祥阳
1. 昆明理工大学医学院疾病和药物遗传学实验室,昆明 650500;2. 昆明理工大学生命科学与技术学院,昆明 650500
性腺母细胞瘤的分子遗传机制研究进展
余莉莉1,董琬如1,2,陈明会1,孔祥阳1
1. 昆明理工大学医学院疾病和药物遗传学实验室,昆明 650500;2. 昆明理工大学生命科学与技术学院,昆明 650500
性腺母细胞瘤(Gonadoblastoma, GB)是一种由性索和生殖细胞演化而来的罕见原位性腺肿瘤,与性腺遗传物质异常有密切联系。80%的GB患者表现为46,XY女性表型,其余为45,XY 和46,XX性别发育异常患者等。35%的GB会进一步演化为无性细胞瘤和精原细胞瘤等恶性肿瘤。由于表型与遗传的异质性,GB的分子遗传机制还未完全揭示。越来越多的研究显示GB的发生与性别分化和决定调控基因(如SRY、WT1、SOX9、Foxl2和TSPY等)之间存在密切关联,且表现出遗传与表观遗传调控相互作用。本文综述了GB的临床表现、病理特征、诊断与治疗措施,总结了性腺遗传异常导致GB的分子遗传与表观遗传调控机制,分析并归纳参与GB形成相关基因的共同表达调控网络,指出了当前研究中的障碍与不足,为进一步研究GB致病分子机制提供新思路。
性腺母细胞瘤;性别发育异常;生殖系统异常;基因调控网络;表观遗传异常
性腺母细胞瘤(Gonadoblastoma,GB)是一类由于性腺遗传异常导致的具有特异性的罕见肿瘤疾病,该病的发生与性别和生殖系统发育异常之间存在着密切的联系[1]。1953年,Scully[2]首次报道了2例固醇类激素分泌型性腺肿瘤的临床症状,并将该病命名为GB。几十年来,科研人员对GB的遗传基础、致病机制以及诊断和治疗措施等方面进行了系统性研究并获得了一系列研究成果[3],如发现和鉴定GB致病基因、完善诊断标准和治疗措施、建立GB发病的病理演化过程模型等。由于GB临床病例分散、遗传和表型异质性较强、相关致病基因复杂等因素,导致开展GB发病分子机制的系统性研究显得十分困难。本课题组一直从事遗传异常导致罕见肿瘤发生的分子机制研究,在前期研究性别和生殖系统发育异常的分子机制时发现GB致病基因如Y染色体性别决定区域(SRY)、Wilms'抑癌基因1(WT1)、SOX9、Foxl2及睾丸特异基因(TSPY)之间存在相互作用关系,表明它们之间可能存在着共同的信号通路和基因相互作用网络。本文通过查阅大量文献并结合本课题组前期研究成果,系统地总结了性腺遗传异常导致GB发生的分子遗传机制,分析并归纳了参与GB发病相关基因共同表达调控网络,指出了当前研究中的障碍和不足,为进一步研究GB致病机制,完善诊断与治疗措施提出合理建议。
1 GB的临床表现、病理特征和诊治措施
GB为罕见性腺肿瘤,多发于青春期前后,主要集中在16~25岁之间[4,5]。患者大部分都表现出性别发育异常,主要的核型为46,XY,此外还有46,XX和45,XY等。流行病学研究显示80%的患者性别表型为女性,仅极少数可成功受孕。部分患者还伴有其他发育异常症状,如Frasier综合征[6]、Turner综合征[7]等。此外,35%的GB会进一步演化为无性细胞瘤和精原细胞瘤等恶性肿瘤[8,9]。
目前,GB的诊断主要依据病理形态特征并综合考察临床病史[10,11]。但是由于GB常伴随其他肿瘤的发生,如生殖细胞肿瘤、两性母细胞瘤和性索瘤等,因此单纯采用病理形态特征不易将GB与其他肿瘤有效区分。此外,通过染色体核型分析对于GB确诊具有重要作用。近几年来,分子诊断技术也广泛应用于GB的诊断上,如研究证实可将性腺组织中β-catenin表达水平作为恶性生殖细胞肿瘤的诊断分子指标,OCT3/4和TSPY蛋白表达水平也可作为评估GB发生风险的生物标志物[12]。Li等[13]指出性腺组织中的TSPY1蛋白水平可用于直接诊断由Y染色体引起的GB。2015年,Van等[14]综合分子生物标志物、遗传多态性、表观遗传变异以及环境因子等风险因子建立了生殖细胞肿瘤发生、诊断和治疗方式的判别模型。
对于表现为Turner综合征的遗传异常性腺患者而言,一般推荐采取预防性的性腺切除措施,而GB的治疗一般采用切除性腺并辅助化疗和放疗等治疗方式。单纯性GB的治疗后康复效果较好,但其中的生殖细胞具有转化为恶性细胞的风险[14];而恶性GB治疗后康复效果差,治疗后存活时间一般在1年左右[15]。
2 GB形成的分子遗传机制
近十几年来,随着分子生物学技术的快速发展,如蛋白印迹、qPCR、基因测序和表观遗传学等研究手段,使得研究人员可以从基因及其表达和翻译等不同分子层次去探讨GB的发病机制。GB的发生与遗传与表观遗传调控之间密切相关。
2.1性别分化和决定相关基因遗传调控异常导致GB的形成
2.1.1SRY基因功能异常导致GB形成
SRY基因位于Y染色体短臂的近侧区,是性别分化的关键开关基因之一,它编码睾丸特异转录因子负责启动睾丸分化和后续的雄性发育[16,17]。SRY蛋白由N-端、C-端和高迁移率簇蛋白域(HMG)组成,HMG的上下游各含有1个核定位信号,分别为nNLS和cNLS,HMG主要与DNA的结合有关,又称为DNA结合模块。临床病例研究发现,GB患者SRY基因存在位点突变、错义突变和SRY基因缺失等多种类型遗传异常(图1),导致SRY基因表达水平下降并通过多级下游表达调控引起雄性支持细胞不能成熟。Shahid等[18]在3位印度46,XY女性患者性腺中发现SRY基因HMG的上游区域密码子57由谷氨酸突变为精氨酸;此外在HMG下游端发现密码子143由丝氨酸突变为半胱氨酸。Helszer等[19]发现46,XY女性患者SRY基因HMG的341位碱基出现错义突变(A>G)。Hersmus等[20]在46,XY女性患者双侧GB中发现nNLS区70位密码子因错义突变导致氨基酸由W突变为L。不仅在GB患者中检测SRY基因存在位点突变和错义突变,而且还发现SRY基因缺失。如Kim等[21]在XXY女性患者样本中发现Y染色体p臂缺失,而Aviv等[22]通过原位荧光杂交实验在性染色体镶嵌现象患者性腺中发现整个Y染色体断臂缺失。此外,Susan等[23]在爱尔兰15岁男孩病例中发现其SRY基因缺失。
与此相反,Bianco等[24]对Turner综合征患者进行基因序列分析后发现在所有16位患者异常性腺中均发现SRY基因的存在。由于Turner综合征具有极高的GB转化风险,所以Bianco等[24]认为遗传异常性腺中Y染色体的存在,特别是SRY基因的存在会增加GB发病的风险。但是该研究仅采用PCR扩增技术检测到SRY基因的存在,并未对SRY基因序列进行分析。
2.1.2WT1突变导致GB形成
WT1基因编码锌指转录因子并具有肿瘤抑制的活性,在肾脏和性腺发育中具有重要作用。WT1基因编码蛋白可以多达24个异构体,这些异构体C-端含有4个锌指结构域,可与DNA和RNA结合,在WT1蛋白的N-端含有抑制结构域、激活结构域和RNA识别模块[25],因此WT1蛋白在下游基因的转录中同时具有激活和抑制转录的双重作用。
WT1基因编码的2个主要的异构体分别为+KTS和−KTS。+KTS在转录中具有重要作用,保证性腺与肾脏的发育,而-KTS则在mRNA修饰等过程中起到重要作用,防止Wilms' 相关肿瘤的发生[26]。Hersmus等[27]研究发现WT1基因内含子9的位点突变会降低+KTS与−KTS的含量比值(< 2:1),导致了部分甚至全部的性腺发育异常并引起GB的形成。此外,研究还表明N-端(外显子1~5)基因突变并未导致性腺与肾脏功能严重不全,因而完整的C-端DNA结合域和正常的+KTS与−KTS含量比值对于性腺与肾脏的发育显得更为重要[27]。大量的研究已经证实在Frasier综合征和GB患者性腺中发现了WT1基因突变和缺失(表1)[6,28~38]。WT1蛋白不同结构域的功能、基因突变与肿瘤之间的相关性见图2。

图1 SR Y蛋白结构图及其在GB中位点突变信息

图2 WT1蛋白不同结构域的功能、基因突变与表型的相关性
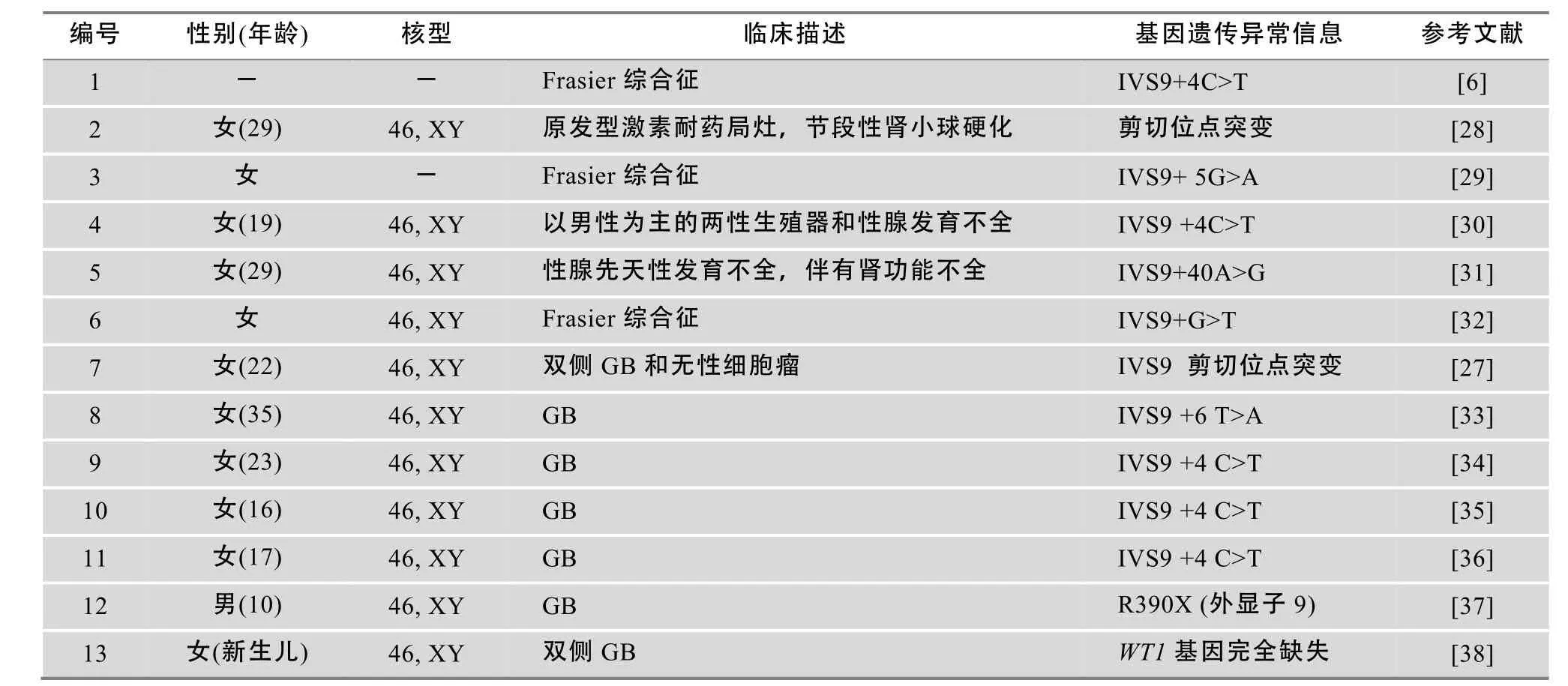
表1 G B及其相关综合征中WT1基因异常情况
2.1.3SOX9与Foxl2基因表达异常诱导GB形成
SOX9基因位于第17号染色体长臂24.3-25.1区段内,是SRY基因的下游基因,该基因含有3个外显子。SOX9蛋白由509个氨基酸组成,由N-端、C-端和HMG组成,C-端含有一个转录激活域,在睾丸发育和支持细胞前体形成,促进支持细胞成熟等方面具有重要的作用[39]。而Foxl2基因是雌性性腺发育的起始基因,对于粒层细胞分化具有重要作用[40]。在基因表达调控上,SOX9基因与Foxl2基因呈现出表达水平相互抑制的特点[40]。此外,有研究指出在46,XY性别发育异常患者性腺中发现在SOX9基因启动子区域存在一个长度为78 kb的区域缺失,表明该区域是性腺转录的增强子[41]。采用免疫组化方法对7例GB样本中SOX9与Foxl2基因表达情况进行了分析,结果发现在性索基质细胞中SOX9基因不表达而Foxl2基因过度表达,在生殖细胞中SOX9与Foxl2基因均不表达,而在GB与无性细胞瘤之间组织中出现Foxl2基因表达逐渐下降。该研究有力地支持了GB发育中性索基质细胞是颗粒细胞起源的假说[42,43]。
2.1.4TSPY作为GB的原癌基因
TSPY基因位于Y染色体的短臂上,转录组长度为1.3 kb,含有6个外显子和5个内含子与胚胎前精原细胞和成年精原细胞以及男性生殖系统中的多能细胞的形成密切相关[44]。人类TSPY基因中含有一个长度为20.4 kb的连续重复单元,重复拷贝数约为35次,该重复单元是TSPY基因的核心区域。早在1987年,Page等[45]就提出在Y染色体上存在一个区域可以促进性别发育异常人群性腺中原位肿瘤形成,并进一步指出该基因在睾丸中发挥正常功能而在异常性腺中作为原癌基因存在。由此,许多学者就这个假设开展了大量的研究,并证实TSPY基因是GB的原癌基因,它不仅在雄性生殖细胞繁殖与分化中起到重要作用,也在早期和晚期GB、睾丸癌中显著表达。TSPY基因不仅在GB组织中过度表达,且在睾丸生殖细胞瘤,如癌变前兆和精原细胞瘤中也过度表达[46]。这些现象表明TSPY在生殖细胞肿瘤发育的早期阶段发挥着重要作用。将不同疾病进程阶段的GB样本进行免疫组化分析,发现外围的滤泡结构均呈现TSPY表达阳性[47]。不仅如此,动物实验也表明TSPY基因过度表达会刺激蛋白合成活性、加速细胞分化和促进肿瘤发育进程[48]。
Skawarn等[49]指出Y染色体性别决定区域基因可能会与TSPY基因的启动子结合并诱导TSPY基因表达,但是该理论一直未得到实验验证。最近,有研究发现TSPY蛋白可以与其自身基因DNA的外显子1结合并显著地增强TSPY基因的转录活性[50],表明TSPY基因可以通过正反馈机制显著增加自身的表达并表现出原癌基因性质(图3)。此外,有学者指出TSPY基因组拷贝数可能存在变异,引起TSPY基因表达量增加并导致GB的形成[51]。
TSPY蛋白存在一个SET/NAP结合域,可以识别SET原癌蛋白和核小体装配蛋白,SET/NAP结合域与周期蛋白B结合,增强周期蛋白B-CDK1激酶活性,加快细胞在G2/M期间转变[52]。SET/NAP结合域还可以与转录延长因子eEF1A结合并增强蛋白合成。以上特性使得TSPY在异常生殖细胞中,如遗传异常的睾丸和子宫中干扰正常的细胞周期调控,引起染色体重排、基因组不稳定并使得未成熟的支持细胞蓄积从而导致肿瘤发生[44]。
2.1.5其他的遗传异常导致GB的形成

图3 TSPY基因作为原癌基因在GB形成中的表达调控机制
除了上述几种导致GB形成的遗传异常外,研究人员还发现一些其他的基因组损伤与GB发生存在密切关系。如Hersmus等[53]发现16例同时患有GB与无精细胞瘤病例受体酪氨酸激酶(c-KIT)基因密码子816存在突变,说明GB的形成与c-KIT突变之间存在一定关联。c-KIT蛋白属于三型酪氨酸激酶受体家族,它同时诱导血小板生长因子受体和巨噬细胞集落刺激受体。c-KIT的配体是干细胞因子和干细胞因子-KIT通路调控子,主要调节生黑色素细胞、血红细胞、肥大细胞、卡哈尔间质细胞、生殖细胞的分化和原生殖细胞的存活。此外,在GB患者中检测到Y染色体中双倍DNA含量,在无性细胞瘤患者中甚至发现等臂染色体12p结构异型[54]。这些都表明GB的发生与Y染色体遗传物质含量与空间结构之间存在密切关系。Y染色体的遗传物质异常,延长了OCT3/4和TSPY基因的表达使得生殖细胞得以存活并导致了GB和无精细胞瘤的形成[55,56]。
2.2性别决定和分化相关基因表观遗传调控异常
导致GB的形成
表观遗传学是针对遗传学而言的,指在不改变基因遗传序列的情况下改变基因表达调控模式,主要包括DNA甲基化、组蛋白修饰和非编码RNA等。相对于遗传异常而言,引起GB形成的表观遗传异常研究显得还十分不足,主要的研究集中在基因组DNA和特异功能基因DNA甲基化异常方面。5-甲基胞嘧啶免疫组化印迹实验揭示胎儿、婴儿与成年人生殖细胞全基因组DNA甲基化水平存在巨大差异,其中未分化的生殖细胞肿瘤(如GB、精原细胞瘤、曲细精管生殖细胞瘤)均表现为基因组DNA低甲基化,而高度分化的生殖细胞瘤(如畸胎瘤、卵黄囊肿瘤和绒膜癌)则表现为基因组DNA高甲基化[57]。在特异功能基因DNA甲基化研究方面,Gimelli等[58]在46,XY女性GB患者病例中发现SRY基因的两个保守区DNA分别表现为低甲基化(甲基化率为2%)和高甲基化(甲基化率为88%),而这种甲基化异常模式会导致SRY基因附近异染色质阻碍模块形成,阻碍了SRY基因与其他转录调控因子的结合从而抑制了SRY基因的表达。不仅如此,Nishino等[59]也在体外实验中证实小鼠性腺发育中SRY基因的转录受到该基因5′端侧链DNA甲基化水平的控制。
尽管到目前为止,GB形成与性别发育和性别决定相关基因表观遗传修饰异常的机制还未阐明,由于在人类性别发育和性别决定的过程中,表观遗传修饰均表现出巨大的改变和精确的调控作用,显示其与GB的形成之间存在潜在且密切的联系[16,60]。
3 基于性别发育基因调控网络的GB形成分子机制
Cool等[4]综合43例GB患者性腺样本的病理切片和基因表达水平特征提出了遗传异常性腺中未分化性腺组织、GB和原位肿瘤的发育模型。该模型指出在胚胎发育阶段,生殖细胞从卵黄囊进入到两性潜能性腺并与支持细胞/粒层细胞混合。在雄性中SRY基因在第6周开始表达并使得支持细胞和生殖细胞在原始性索中发生组合,在SRY基因下游的其他雄性性别决定基因的作用下,这些性索分化成曲细精管。但是在病理条件下正常生殖细胞发育会受阻或延迟,因而会增加曲细精管内的生殖细胞肿瘤的发生风险。在遗传异常性腺中,SRY基因的低表达或不表达,其他雄性决定基因的异常表达均会抑制性索形成和进一步分化。因而,减数分裂通路发育也会受到阻碍,导致形成持续的未分化性腺组织。在未分化的性腺组织和未成熟的性索中存活的生殖细胞具有转化为GB的高风险。然而该模型并没有很好地将组织形态学与基因表达调控网络的联系有机地整合在一起,以便系统地揭示GB的致病分子机制[2]。
要解析GB的致病分子机制需要考虑以下6点因素:(1)在人类性别分化和决定过程中,基因表达与信号传递过程均受到遗传与表观遗传调控的影响;(2)人类怀孕6周后SRY基因开始表达并引起性二态形成[61];(3)GB患者的核型与表型主要表现为46,XY女性[4],且GB均形成在发育异常性腺中;(4)临床样本研究显示GB的形成与胚胎性腺发育和分化过程相关基因,如SRY、WT1、SOX9、Foxl2和TSPY等基因密切相关[3,62,63];(5)在遗传异常性腺上,TSPY基因表达水平显著增高且表达周期延长,扰乱细胞正常增殖周期,且在滤泡中充满TSPY-阳性肿瘤生殖细胞[64];(6)未见文献报道GB患者性腺中TSPY基因异常,如基因突变、倒置、缺失、基因关键域重复等。综合文献资料和本课题组前期研究结果,我们总结出一个基于性别发育基因调控网络的GB形成机制的假说:以SRY基因表达为核心的性腺细胞分化基因表达调控网络与以TSPY基因为核心的生殖系统细胞增殖基因表达调控机制的异常导致了GB的形成。
在性腺发育过程中,转录因子,如GATA4、WT1、SF1等,直接或者间接作为信号传导途径因子激活SRY基因的表达。而性二态性则由Y染色体编码的SRY激活并引起下游SOX9基因的表达促进睾丸的发育。与此同时,SOX9基因与Foxl2基因经过β-catenin与WNT4因子的相互传导而表达相互抑制,从而分别促进雄性与雌性生殖系统的发育[56]。在人类雄性性别分化与决定机制中,TSPY基因表达存在两种调控机制:(1)SRY基因与TSPY基因表达之间相互应答的机制,即SRY基因的正常表达激活了TSPY基因的正常表达,促进了支持细胞的成熟和生殖系统的发育。通过文献对比,SRY基因与TSPY基因的表达均应受到表观遗传机制调控。由于在胚胎发育早期阶段,存在一系列重大的表观遗传事件,如DNA甲基化和去甲基化等,所以SRY与TSPY基因均受到表观遗传调控机制的影响[58,59]。(2)未成熟的支持细胞的积累反馈促进了TSPY基因的表达,且随着未成熟的支持细胞积累增加,这个反馈促进作用增强。由于染色体水平、基因水平和表观遗传调控异常等导致SRY基因表达异常下调,使得支持细胞前体无法进一步发育形成成熟的支持细胞,而增强和延长了TSPY基因的表达,促进了细胞的增殖,并激活了GB的形成(图4)。
4 结语与展望
GB是特异且罕见的由性别与生殖系统发育异常引起的肿瘤。虽然科研人员对该疾病的临床病理,分子机制和诊治措施等方面进行了大量的研究,得到了一系列重要认识,但是由于直接病例样本少,发病机制复杂且研究分散,关于该病发生的机制还有许多问题有待进一步研究。第一,GB的发病史仍不清楚,临床表现上患者通常表现为女性,但存在一定程度的男性化特征。诊断上,原发性闭经并伴随Swyers综合症常被诊断为GB。临床研究证实,除Swyer综合症(46,XY)外,雄激素不敏感症(46,XY)、混合型性腺遗传异常(45,X/46,XY)、Turner综合症、Frasier综合症、Denys-Drash综合症、9p部分单染色体等均与GB的发生风险密切关联[65,66]。第二,环境和营养与表观遗传调控相互作用在GB形成中具有重要作用,特别是患者母亲孕期生活史,因而开展GB患者,特别是散发性病例生活史与表观遗传调控的研究对于揭示GB形成机制具有重要意义。第三,GB形成的可能分子表达调控机制还不清楚。尽管Page假设GB的发展依赖于Y染色体的一部分,称为GBY染色体域[45],然而,有报道发现GB患者具有46,XX基因型,显示存在Y染色体以外的分子机制导致GB的发生[67]。到目前为止,研究已经发现并鉴定的GB发生的基因,包括WT1、SRY、SF1、SOX9、Foxl2等,但其他与性别发育相关的基因还未见研究报道。第四,肿瘤的发生是多因素共同作用的结果,特别是性别分化和生殖系统发育调控基因与GBY染色体域基因之间的相互作用的研究目前还未见报道[68]。第五,GB是无性细胞瘤发生的前兆,但是GB的转移和侵入机制仍未阐明[68,69],因而无法准确评估其转化为恶性肿瘤的风险。目前对GB患者采取的主要预防措施是早期切除,导致了治疗方式简单粗糙和切除滥用。
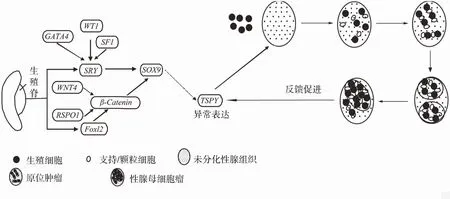
图4 基于性别发育基因调控网络的GB形成的可能模型
为了更加全面地了解GB形成规律,我们认为在开展有限且分散的GB病例研究时需要将患者本人及其家族成员的疾病史和生活史、异常性腺的病理形态特征、性别发育调控网络相关基因的遗传物质基础及表达水平等进行系统研究,在报道GB病例研究成果时详细描述临床特征,增强研究数据之间的对比性。通过对数据的综合分析更好地揭示GB形成机制,进一步完善GB的诊断技术和治疗方案。
[1] Cools M. Germ cell cancer risk in DSD patients. Ann d'Endocrinol, 2014, 75(2): 67-71.
[2] Scully RE. Gonadoblastoma. A gonadal tumor related to the dysgerminoma (seminoma) and capable of sex-hormone production. Cancer, 1953, 6(3): 455-463.
[3] Ulbright TM, Young RH. Gonadoblastoma and selected other aspects of gonadal pathology in young patients with disorders of sex development. Semin Diagn Pathol, 2014,31(5): 427-440.
[4] Cools M, Stoop H, Kersemaekers AMF, Drop SLS, Wolffenbuttel KP, Bourguignon JP, Slowikowska-Hilczer J,Kula K, Faradz SMH, Oosterhuis JW, Looijenga LHJ. Gonadoblastoma arising in undifferentiated gonadal tissue within dysgenetic gonads. J Clin Endocrinol Metab, 2006,91(6): 2404-2413.
[5] Cools M, Looijenga LHJ, Wolffenbuttel KP, T'Sjoen G. Managing the risk of germ cell tumourigenesis in disorders of sex development patients. Endocr Dev, 2014, 27:185-196
[6] Kitsiou-Tzeli S, Deligiorgi M, Malaktari-Skarantavou S,Vlachopoulos C, Megremis S, Fylaktou I, Traeger-Synodinos J, Kanaka-Gantenbein C, Stefanadis C, Kanavakis E. Sertoli cell tumor and gonadoblastoma in an untreated 29-year-old 46, XY phenotypic male with Frasier syndrome carrying a WT1 IVS9+4C>T mutation. Hormones, 2012, 11(3): 361-367.
[7] Gómez Llorente JL, Hernández IA, Muñoz Sánchez MD,de Cabo JM, Perales AB. Early presentation of bilateral gonadoblastoma in Turner syndrome. J Pediatr Hematol Oncol, 2013, 35(3): 240-241.
[8] Du X, Zhang XH, Li YM, Han YK. 46, XY female sex reversal syndrome with bilateral gonadoblastoma and dysgerminoma. Exp Ther Med, 2014, 8(4): 1102-1104.
[9] Kao CS, Ulbright TM, Idrees MT. Gonadoblastoma: an immunohistochemical study and comparison to Sertoli cell nodule with intratubular germ cell neoplasia, with pathogenetic implications. Histopathology, 2014, 65(6):861-867.
[10] Ulbright TM. Gonadoblastoma and hepatoid and endometrioid-like yolk sac tumor: an update. Int J Gynecol Pathol,2014, 33(4): 365-373.
[11] Gorosito M, Pancera B, Sarancone S, Nocito AL. Gonadoblastoma: an unusual ovarian tumor. Ann Diagn Pathol,2010, 14(4): 247-250.
[12] Palma I, Garibay N, Pena-Yolanda R, Contreras A, Raya A,Dominguez C, Romero M, Aristi G, Queipo G. Utility of OCT3/4, TSPY and β-catenin as biological markers for gonadoblastoma formation and malignant germ cell tumor development in dysgenetic gonads. Dis Markers, 2013,34(6): 419-424.
[13] Li S, Mo CJ, Huang S, Yang S, Lu Y, Peng QL, Wang J,Deng Y, Qin X, Liu YK. Over-expressed Testis-specific Protein Y-encoded 1 as a novel biomarker for male hepatocellular carcinoma. PLoS One, 2014, 9(2): e89219.
[14] Van der Zwan YG, Biermann K, Wolffenbuttel KP, Cools M, Looijenga LHJ. Gonadal maldevelopment as risk factor for germ cell cancer: towards a clinical decision model. Eur Urol, 2015, 67(4): 692-701.
[15] Peña-Alonso R, Nieto K, Alvarez R, Palma I, Nájera N,Eraña L, Dorantes LM, Kofman-Alfaro S, Queipo G. Distribution of Y-chromosome-bearing cells in gonadoblastoma and dysgenetic testis in 45, X/46, XY infants. Mod Pathol, 2005, 18(3): 439-445.
[16] Larney C, Bailey TL, Koopman P. Switching on sex:transcriptional regulation of the testis-determining gene Sry. Development, 2014, 141(11): 2195-2205.
[17] 裴开颜, 王介东. Y染色体性别决定区(SRY): 性别决定关键开关. 生殖医学杂志, 2012, 21(4): 400-405.
[18] Shahid M, Dhillion VS, Jain N, Hedau S, Diwakar S,Sachdeva P, Batra S, Das BC, Husain SA. Two new novel point mutations localized upstream and downstream of the HMG box region of the SRY gene in three Indian 46, XY females with sex reversal and gonadal tumour formation. Mol Hum Reprod, 2004, 10(7): 521-526.
[19] Helszer Z, Dmochowska A, Szemraj J, Słowikowska-Hilczer J, Wieczorek M, Jędrzejczyk S, Kałużewski B. A novel mutation (c. 341A>G) in the SRY gene in a 46, XY female patient with gonadal dysgenesis. Gene,2013, 526(2): 467-470.
[20] Hersmus R, de Leeuw BH, Stoop H, Bernard P, van Doorn HC, Brüggenwirth HT, Drop SL, Oosterhuis JW, Harley VR, Looijenga LH. A novel SRY missense mutation af-fecting nuclear import in a 46, XY female patient with bilateral gonadoblastoma. Eur J Hum Genet, 2009, 17(2):1642-1649.
[21] Kim J, Kim SH, Song J, Choi JR, Kim HS, Lee KA. An SRY-deleted XXY female resulting from a paternally inherited t(Y;22). Ann Clin Lab Sci, 2010, 40(3): 295-299.
[22] Aviv H, Heller D, Fajardo A, Hoot A, Mavaro L. Sex chromosome mosaicism in gonads of a fetus with cystic hygroma and deletion of the short arm of Y chromosome including loss of SRY. Am J Med Genet,2001, 102(2): 157-160.
[23] O'Connell SM, Lynch SA, Coyle D, McDermott M,O'Sullivan M, Roche E, Quinn F, Cody D. The incidence of childhood gonadoblastoma over 15 years in the Republic of Ireland. ESPE Abstracts, 2014, 1(2): 211.
[24] Bianco B, Lipay M, Guedes A, Oliveira K, Verreschi ITN. SRY gene increases the risk of developing gonadoblastoma and/or nontumoral gonadal lesions in Turner syndrome. Int J Gynecol Pathol, 2009, 28(2): 197-202.
[25] Lindstedt I, Lindgren MA, Andersson E, Engstrom W. The WT1 gene-its role in tumourigenesis and prospects for immunotherapeutic advances. In Vivo, 2014, 28(5):675-681.
[26] Bagchi D, Andrade J, Shupnik MA. A new role for wilms tumor protein 1: differential activities of + KTS and -KTS variants to regulate LHβ transcription. PLoS One, 2015,10(1): e0116825.
[27] Hersmus R, van der Zwan YG, Stoop H, Bernard P, Sreenivasan R, Oosterhuis JW, Brüggenwirth HT, de Boer S,White S, Wolffenbuttel KP, Alders M, McElreavy K, Drop SLS, Harley VR, Looijenga LH. A 46, XY female DSD patient with bilateral gonadoblastoma, a novel SRY missense mutation combined with a WT1 KTS splice-site mutation. PLoS One, 2012, 7(7): e40858.
[28] Denamur E, Bocquet N, Baudouin V, Da Silva F, Veitia R,Peuchmaur M, Elion J, Gubler MC, Fellous M, Niaudet P,Loirat C. WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental glomerulosclerosis. Kidney Int, 2000, 57(5): 1868-1872.
[29] Shimoyama H, Nakajima M, Naka H, Park YD, Hori K,Morikawa H, Yoshioka A. A girl with bilateral ovarian tumours: Frasier syndrome. Eur J Pediatr, 2002, 161(2):81-83.
[30] Melo KFS, Martin RM, Costa EMF, Carvalho FM, Jorge AA, Arnhold IJP, Mendonca BB. An unusual phenotype of Frasier syndrome due to IVS9 +4C>T mutation in the WT1 gene: predominantly male ambiguous genitalia and absence of gonadal dysgenesis. J Clin Endocrinol Metab, 2002, 87(6): 2500-2505.
[31] Pérez de Nanclares G, Castaño L, Bilbao JR, Vallo A, Rica I, Vela A, Martul P. Molecular analysis of Frasier syndrome: mutation in the WT1 gene in a girl with gonadal dysgenesis and nephronophthisis. J Pediatr Endocrinol Metab, 2002, 15(7): 1047-1050.
[32] Subbiah V, Huff V, Wolff JEA, Ketonen L, Lang FF Jr,Stewart J, Langford L, Herzog CE. Bilateral gonadoblastoma with dysgerminoma and pilocytic astrocytoma with WT1 GT-IVS9 mutation: A 46 XY phenotypic female with Frasier syndrome. Pediatr Blood Cancer, 2009, 53(7):1349-1351.
[33] Barbaux S, Niaudet P, Gubler MC, Grünfeld JP, Jaubert F,Kuttenn F, Fékété CN, Souleyreau-Therville N, Thibaud E,Fellous M, McElreavey K. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet,1997, 17(4): 467-470.
[34] Okuhara K, Tajima S, Nakae J, Sasaki S, Tochimaru H,Abe S, Fujieda K. A Japanese case with Frasier syndrome caused by the splice junction mutation of WT1 gene. Endocr J, 1999, 46(5): 639-642.
[35] Kikuchi H, Takata A, Akasaka Y, Fukuzawa R, Yoneyama H, Kurosawa Y, Honda M, Kamiyama Y, Hata J. Do intronic mutations affecting splicing of WT1 exon 9 cause Frasier syndrome? J Med Genet, 1998, 35(1): 45-48.
[36] Bönte A, Schröder W, Denamur E, Querfeld U. Absent pubertal development in a child with chronic renal failure:the case of Frasier syndrome. Nephrol Dial Transplant,2000, 15(10): 1688-1690.
[37] Kohsaka T, Tagawa M, Takekoshi Y, Yanagisawa H, Tadokoro K, Yamada M. Exon 9 mutations in the WT1 gene,without influencing KTS splice isoforms, are also responsible for Frasier syndrome. Hum Mutat, 1999, 14(6):466-470.
[38] Finken MJ, Hendriks YM, van der Voorn JP, Veening MA,Lombardi MP, Rotteveel J. WT1 deletion leading to severe 46, XY gonadal dysgenesis, Wilms tumor and gonadoblastoma: case report. Horm Res Paediatr, 2015, 83(3):211-216.
[39] Jo A, Denduluri S, Zhang BS, Wang ZL, Yin LJ, Yan ZJ,Kang R, Shi LL, Mok J, Lee MJ, Haydon RC. The versatile functions of Sox9 in development, stem cells, and human diseases. Genes Dis, 2014, 1(2): 149-161.
[40] Jaubert F, Galmiche L, Lortat-Jacob S, Fournet JC, Fellous M. Foxl-2 in gonad development and pathology. Arkh Patol, 2011, 73(4): 10-13.
[41] Vetro A, Ciccone R, Giorda R, Patricelli MG, Della Mina E, Forlino A, Zuffardi O. XX males SRY negative: a con-firmed cause of infertility. J Med Genet, 2011,48(10): 710-712.
[42] Hersmus R, Kalfa N, de Leeuw B, Stoop H, Oosterhuis JW, de Krijger R, Wolffenbuttel KP, Drop SLS, Veitia RA,Fellous M, Jaubert F, Looijenga LHJ. FOXL2 and SOX9 as parameters of female and male gonadal differentiation in patients with various forms of disorders of sex development (DSD). J Pathol, 2008, 215(1): 31-38.
[43] Buell-Gutbrod R, Ivanovic M, Montag A, Lengyel E, Fadare O, Gwin K. FOXL2 and SOX9 distinguish the lineage of the sex cord-stromal cells in gonadoblastomas. Pediatr Dev Pathol, 2011, 14(5): 391-395.
[44] Lau YFC. Gonadoblastoma, testicular and prostate cancers,and the TSPY gene. Am J Hum Genet, 1999, 64(4):921-927.
[45] Page DC. Hypothesis: a Y-chromosomal gene causes gonadoblastoma in dysgenetic gonads. Development, 1987,101(Suppl.): 151-155.
[46] Kersemaekers AMF, Honecker F, Stoop H, Cools M,Molier M, Wolffenbuttel K, Bokemeyer C, Li YM, Lau YFC, Oosterhuis JW, Looijenga LHJ. Identification of germ cells at risk for neoplastic transformation in gonadoblastoma: an immunohistochemical study for OCT3/4 and TSPY. Hum Pathol, 2005, 36(5): 512-521.
[47] Li YM, Vilain E, Conte F, Rajpert-De Meyts E, Lau YFC. Testis-specific protein Y-encoded gene is expressed in early and late stages of gonadoblastoma and testicular carcinoma in situ. Urol Oncol, 2007, 25(2): 141-146.
[48] Schubert S, Schmidtke J. Transgenic mouse studies to understand the regulation, expression and function of the testis-specific protein Y-encoded (TSPY) gene. Genes,2010, 1(2): 244-262.
[49] Skawran B, Schubert S, Dechend F, Vervoorts J, Nayernia K, Lüscher B, Schmidtke J. Characterization of a human TSPY promoter. Mol Cell Biochem, 2005, 276(1-2):159-167.
[50] Kido T, Lau YFC. The Y-located gonadoblastoma gene TSPY amplifies its own expression through a positive feedback loop in prostate cancer cells. Biochem Biophys Res Commun, 2014, 446(1): 206-211.
[51] Shen Y, Yan YL, Liu YQ, Zhang SZ, Yang D, Zhang P, Li L, Wang Y, Ma YX, Tao DC, Yang Y. A significant effect of the TSPY1 copy number on spermatogenesis efficiency and the phenotypic expression of the gr/gr deletion. Hum Mol Genet, 2013, 22(8): 1679-1695.
[52] Kandalaft LE, Zudaire E, Portal-Núñez S, Cuttitta F, Jakowlew SB. Differentially expressed nucleolar transforming growth factor-β1 target (DENTT) exhibits an inhibitory role on tumorigenesis. Carcinogenesis, 2008,29(6): 1282-1289.
[53] Hersmus R, Stoop H, van de Geijn GJ, Eini R, Biermann K, Oosterhuis JW, DHooge C, Schneider DT, Meijssen IC,Dinjens WNM, Dubbink HJ, Drop SLS, Looijenga LHJ. Prevalence of c-KIT mutations in gonadoblastoma and dysgerminomas of patients with disorders of sex development (DSD) and ovarian dysgerminomas. PLoS One,2012, 7(8): e43952.
[54] Changchien YC, Haltrich I, Micsik T, Kiss E, Fónyad L,Papp G, Sápi Z. Gonadoblastoma: Case report of two young patients with isochromosome 12p found in the dysgerminoma overgrowth component in one case. Pathol Res Pract, 2012, 208(10): 628-632.
[55] Hersmus R, Stoop H, Turbitt E, Oosterhuis JW, Drop SL,Sinclair AH, White SJ, Looijenga LH. SRY mutation analysis by next generation (deep) sequencing in a cohort of chromosomal Disorders of Sex Development (DSD)patients with a mosaic karyotype. BMC Med Genet, 2012,13: 108.
[56] Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol, 2013,9(2): 79-91.
[57] Wermann H, Stoop H, Gillis AJ, Honecker F, van Gurp RJ,Ammerpohl O, Richter J, Oosterhuis JW, Bokemeyer C,Looijenga LH. Global DNA methylation in fetal human germ cells and germ cell tumours: association with differentiation and cisplatin resistance. J Pathol, 2010; 221(4):433-442.
[58] Gimelli G, Giorda R, Beri S, Gimelli S, Zuffardi O. A 46,X, inv(Y) young woman with gonadal dysgenesis and gonadoblastoma: Cytogenetics, molecular, and methylation studies. Am J Med Genet A, 2006, 140A(1): 40-45.
[59] Nishino K, Hattori N, Tanaka S, Shiota K. DNA methylation-mediated control of Sry gene expression in mouse gonadal development. J Biol Chem, 2004, 279(21):22306-22313.
[60] Tachibana M. Epigenetic regulation of mammalian sex determination. J Med Invest, 2015, 62: 19-23.
[61] Wilhelm D, Martinson F, Bradford S, Wilson MJ, Combes AN, Beverdam A, Bowles J, Mizusaki H, Koopman P. Sertoli cell differentiation is induced both cell-autonomously and through prostaglandin signaling during mammalian sex determination. Dev Biol, 2005, 287(1): 111-124.
[62] 程汉华, 周荣家. 胚胎早期的发育选择: 性别决定. 遗传, 2007, 29(2): 145-149.
[63] 李萌, 贺竹梅. 遗传学教学中性别决定关键基因的阐述.遗传, 2014, 36(6): 611-617.
[64] Li YM, Tabatabai ZL, Lee TL, Hatakeyama S, Ohyama C,Chan WY, Looijenga LHJ, Lau YFC. The Y-encoded TSPY protein: a significant marker potentially plays a role in the pathogenesis of testicular germ cell tumors. Hum Pathol, 2007, 38(10): 1470-1481.
[65] Patel PR, Pappas J, Arva NC, Franklin B, Brar PC. Early presentation of bilateral gonadoblastomas in a Denys-Drash syndrome patient: a cautionary tale for prophylactic gonadectomy. J Pediatr Endocrinol Metab,2013; 26(9-10): 971-974.
[66] Quinonez SC, Park JM, Rabah R, Owens KM, Yashar BM,Glover TW, Keegan CE. 9p partial monosomy and disorders of sex development: review and postulation of a pathogenetic mechanism. Am J Med Genet A, 2013,161A(8): 1882-1896.
[67] Esin S, Baser E, Kucukozkan T, Magden HA. Ovarian gonadoblastoma with dysgerminoma in a 15-year-old girl with 46, XX karyotype: case report and review of the literature. Arch Gynecol Obstet, 2012, 285(2): 447-451.
[68] Bai ST, Wei S, Ziober A, Yao Y, Bing ZY. Expression of SALL4 and SF-1 in gonadoblastoma: useful markers in the identification of the invasive germ cell component. Int J Gynecol Pathol, 2013, 32(4): 379-383.
[69] Morel Y, Roucher F, Mallet D, Plotton I. Genetic of gonadal determination. Ann Endocrinol, 2014, 75(2):32-39.
(责任编委: 方向东)
Progress in the molecular genetic mechanism of gonadoblastoma
Lili Yu1, Wanru Dong1,2, Minghui Chen1, Xiangyang Kong1
1. Department of Medical Genetics and Pharmacogenomics, Faculty of Medicine, Kunming University of Science and Technology,Kunming 650500, China;
2. College of Life Science and Technology ,Faculty of Medicine, Kunming University of Science and Technology, Kunming 650500,China
Gonadoblastoma (GB), a rare in situ germ cell tumor derived from sex cord and germ cells, is closely associated with gonadal dysgenesis. About 80% of GB individuals exhibit 46, XY female phenotype while the others are 45, XY and 46, XX with disorders of sex development. Moreover, 35% of GB can eventually develop into malignant tumors, such as seminoma and dysgerminoma tumors. The molecular genetic mechanism of GB remains to be fully uncovered due to phenotypic and genetic heterogeneity. Increasing studies show that the formation of GB is closely related to genes regulating sexual differentiation and determination (e.g., SRY, WT1, SOX9, Foxl2, TSPY,etc), and is affected by the interaction of genetic and epigenetic regulation. Here we describe the clinical and pathological features, diagnosis and treatment of GB, and also summarize the molecular genetic and epigenetic mechanisms underlying the gonadal abnormalities that lead to GB. We analyze and construct the common gene regulatory networks related to the development of GB, and describe some obstacles and deficiencies in current studies to provide innovativeperspectives on further studying the pathological and molecular mechanisms of GB.
gonadoblastoma; disorders of sex development; gonadal abnormalities; gene regulatory network; epigenetic regulation
2015-03-25;
2015-07-24
余莉莉,硕士研究生,专业方向:遗传学。 E-mail: yonily6@163.com
孔祥阳,博士,教授,研究方向:疾病和药物遗传学。 E-mail: kxy02@kmust.edu.cn
10.16288/j.yczz.15-124
网络出版时间: 2015-8-715:18:35
URL: http://www.cnki.net/kcms/detail/11.1913.R.20150807.1518.002.html
猜你喜欢
解放军医学院学报(2022年9期)2022-11-06
文萃报·周五版(2022年24期)2022-06-21
湖泊科学(2020年4期)2020-07-17
中国医学影像技术(2019年10期)2019-10-24
河北渔业(2019年7期)2019-08-27
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
中国男科学杂志(2016年9期)2016-03-20
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
