
过氧化物酶体增殖物激活受体γ辅激活物1-α在急性肾损伤中的作用
2018-08-31 09:50张梦希,董伟,综述等
肾脏病与透析肾移植杂志 2018年4期
急性肾损伤(AKI)是威胁人类健康的常见危重病,其发病机制复杂,至今尚未完全阐明。目前临床尚缺乏预防和治疗AKI的有效药物,防治AKI已成全球范围内重大公共卫生问题。据统计,约7%~18%住院患者会发生AKI[1],而ICU重症患者中,AKI发生率更是高达38%~51%[2]。
肾脏是人体第二大能量需求器官,其线粒体含量仅次于心脏。在肾小管上皮细胞,尤其是近端小管和髓袢升支粗段细胞中线粒体含量十分丰富。线粒体通过其内膜电子传递链将质子从线粒体基质泵入膜间隙,在内膜上产生电化学梯度,从而驱动二磷酸腺苷(ADP)磷酸化为三磷酸腺苷(ATP),用来维持肾小管上皮细胞的电化学梯度及协助溶质运输,以实现肾脏重吸收等重要生理功能。既往研究发现,过氧化物酶体增殖物激活受体γ辅激活物1-α(peroxisome proliferator-activated receptor γ coactivator 1-α,PGC1-α)是维持线粒体正常能量代谢、形态及线粒体生物合成的关键调控因子,主要表达于心、肾、骨骼肌等能量要求高和线粒体丰富的组织器官,在线粒体生成、氧化磷酸化及脂肪酸氧化等一系列能量代谢过程中发挥重要作用。本文主要简述PGC1-α在AKI的发生发展中可能发挥的重要作用。
AKI时肾小管上皮细胞线粒体损伤
线粒体被誉为“细胞的动力车间”,是能量代谢和氧化应激的调控中枢。肾小管上皮细胞线粒体功能障碍是AKI的重要病理生理改变,由此引起的能量代谢异常,释放活性氧(reactive oxygen species,ROS)、细胞色素c、凋亡激活因子等所致的肾小管上皮细胞程序性死亡是重症AKI的关键环节[3]。Brooks等[4]在C57BL/6小鼠模拟肾缺血再灌注损伤(ischemical reperfusion injury,IRI),3D电镜扫描见肾小管上皮细胞内线粒体完全碎裂成点状亚细胞器结构。在脓毒症患者尸检、缺血性AKI肾脏活检组织中,电镜下均可见近端肾小管上皮细胞线粒体数量减少、肿胀、空泡形成、线粒体嵴排列紊乱、溶解甚至断裂[5-6]。由此可见,多种病因所致AKI均存在肾小管上皮细胞的线粒体损伤。
AKI时肾小管上皮细胞线粒体除了形态结构改变外,还存在功能改变。首先,AKI时肾小管上皮细胞线粒体电子传递受到抑制,导致质子漏出和ROS增多,ROS可进一步加重线粒体损伤,引发肾小管上皮细胞能量代谢紊乱[7]。其次,线粒体ATP产生减少,导致细胞内离子转运失衡、脂质和代谢物堆积[7]。而线粒体生物合成在AKI肾功能恢复中起着重要作用[9]。因此,预防线粒体损伤对于减轻AKI具有重要意义,开发针对肾小管上皮细胞线粒体功能障碍或促进线粒体再生的新疗法,以控制AKI和促进受损肾组织修复具有广阔前景。
PGC1-α与线粒体损伤
哺乳动物线粒体内约有1 500种蛋白,除少数蛋白由线粒体DNA编码合成,其他大多数由核DNA(nDNA)编码。近年来,PGC1家族被认为是线粒体稳态最重要的调控者[10]。该家族由三个成员组成,即PGC1-α、PGC1-β和PGC相关辅激活因(PRC),三者结构高度同源,作为核转录辅激活因子与转录因子及启动子相互作用,发挥生物学功能[11]。其中,目前研究最为深入的是PGC1-α。PGC1-α由PPARGC1A基因编码,该基因定位于染色体4p15.2,全长约67 kb,由13个外显子组成,编码798个氨基酸组成的蛋白质(分子量91 kD),在人和小鼠间高度保守。大量研究表明,PGC1-α在糖异生和葡萄糖转运、脂肪酸氧化、ROS抑制、线粒体生物合成和氧化磷酸化等方面发挥重要调控作用[12]。
作为辅转录因子,PGC1-α连接转录因子和启动子,增强转录因子对下游线粒体相关蛋白转录调控。PGC1-α可通过辅助核红系因子(Nrf)、雌激素相关受体α(estrogen-related receptor-alpha,ERR-α)等转录因子,参与线粒体氧化磷酸化、营养物质代谢及维持线粒体稳态[13-14]。此外,PGC1-α可促进抗氧化酶的表达来抑制ROS活性,下调PGC1-α可致线粒体膜电势降低,伴ROS爆发和蛋白氧化损伤加剧,从而破坏线粒体稳态[15]。
PGC1-α活性受转录调控及翻译后修饰调节。PGC1-α受肌细胞增强子2(myocyte enhancer factor,MEF2)、叉头框家族(forkhead transcription factor,FOXO)、肝细胞核因子1β(hepatocyte nuclear factor 1β,HNF1β)等转录因子调控[15]。PGC1-α翻译后修饰调节包括磷酸化、乙酰化、甲基化、泛素化、小泛素样修饰等。其中磷酸化、乙酰化是最重要的修饰方式。AMPK,p38MAPK,Akt是磷酸化PGC1-α的三种经典蛋白激酶,可提高其转录活性、增强分子稳定性、延长降解半衰期。此外还有糖原合成激酶3β(glycogen synthase kinase 3β,GSK3β)、Cdc2样激酶2(Cdc2-like kinase 2,Clk2)等也可使PGC1-α磷酸化[17-18]。乙酰基转移酶GCN5可乙酰化PGC-1α赖氨酸残基,导致PGC1-α转录活性丧失,继而从靶基因启动子区脱离[19];而去乙酰化修饰主要由沉默调节因子(SIRT)介导,当氧化态/还原态烟酰胺腺嘌呤二核苷酸(NAD+/ NADH)升高时,SIRT可使PGC1-α去乙酰化从而提高其转录活性(图1)[20]。
PGC1-α在AKI中的研究进展
最早在顺铂诱导的小鼠AKI模型中可见肾近端小管细胞PGC1-α转录及表达均下降,伴随着线粒体功能、结构紊乱及再生障碍[21]。这一现象在缺血再灌注AKI模型、脓毒症性AKI模型中均得到证实[13,22]。由此可见,AKI时普遍存在肾小管上皮细胞PGC1-α下调。随后,用过氧化氢(H2O2)诱导近端小管细胞氧化应激,发现过表达PGC1-α可促进氧化应激后线粒体数目和功能恢复,提高细胞存活率[23]。此外,Tran等[22]发现虽然普通小鼠在PGC1-α敲除后,线粒体没有明显的超微结构缺陷,肾功能正常,但脓毒症性AKI小鼠敲除PGC1-α后较野生型小鼠肾损伤程度更严重且持续存在,而过表达PGC1-α可增加近端小管细胞线粒体数量、提高呼吸链功能、恢复细胞氧耗及下游靶基因表达,减轻AKI。这些研究结果提示肾小管上皮细胞PGC1-α的下调介导了AKI,并且PGC1-α对于AKI肾功能恢复具有重要作用。
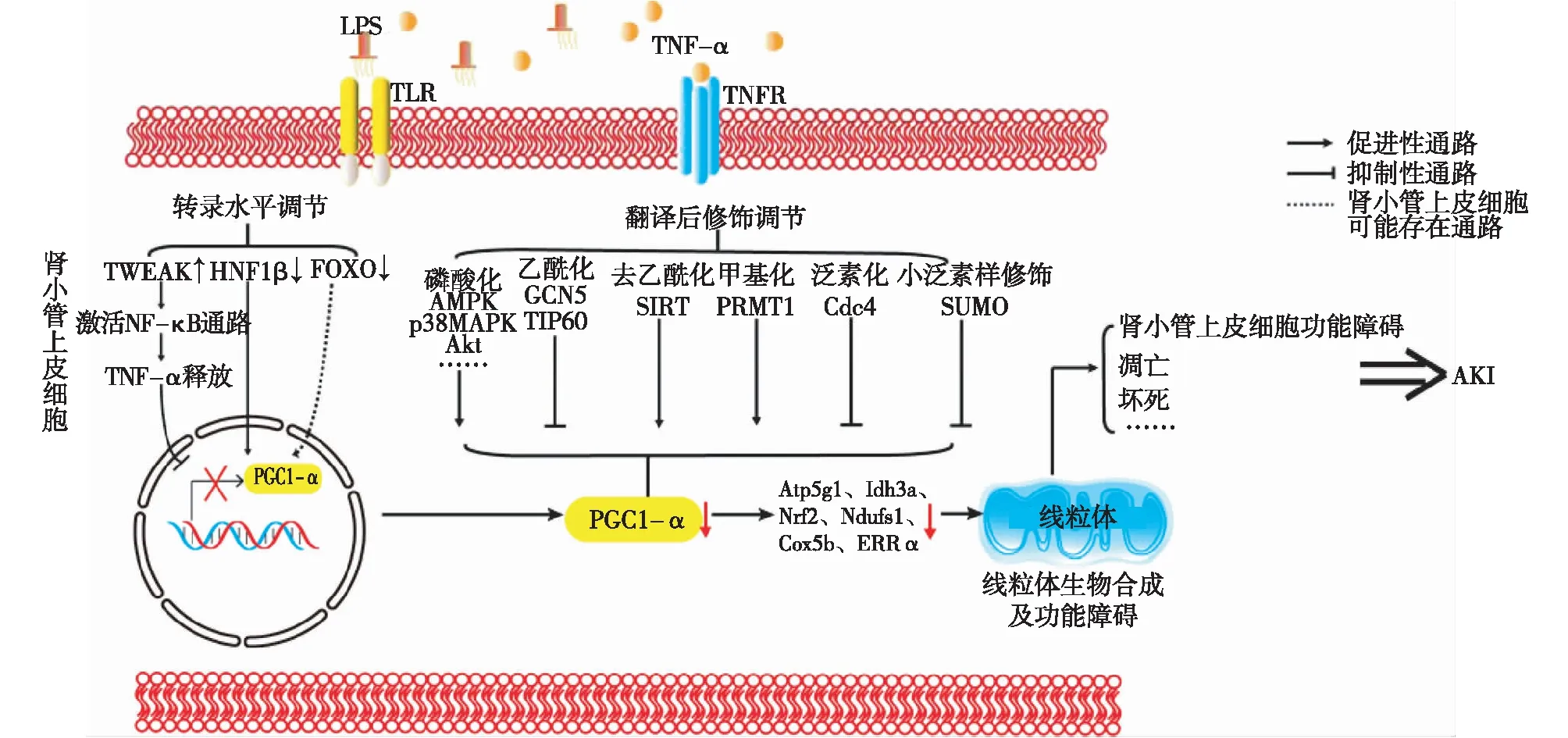
图1 PGC1-α的调节及其在脓毒症性AKI中的作用机制
那么,PGC1-α通过哪些途径影响AKI?NAD是细胞内重要的脱氢酶辅酶,连接三羧酸循环和呼吸链。Tran等[24]进一步研究发现,缺血再灌注AKI时,敲除PGC1-α加重了肾脏局部NAD前体缺乏、近端小管细胞脂质堆积,导致肾损伤持续加重;而PGC1-α过表达可促进NAD从头合成关键酶的表达、前列腺素PGE2及脂肪分解产物β羟基丁酸生成,从而升高肾小管上皮细胞内NAD水平、促进脂质代谢、逆转线粒体功能紊乱,进而改善肾脏缺血再灌注损伤。另外,Choi等[13]发现PGC1-α的细胞保护作用与上调Nrf2有关,PGC1-α过表达可活化p38/GSK3β/Nrf2轴,上调Nrf2表达,抑制H2O2诱导的HK-2细胞凋亡,而Nrf2特异性基因敲除可阻断该过程。随后他们在缺血再灌注损伤小鼠模型中验证了该观点。由此证实PGC1-α可通过上调Nrf2来减轻AKI。
AKI时PGC1-α下调的原因是什么呢?Tran等[22]发现脓毒症性AKI中PGC1-α mRNA明显减少。因此推测,AKI时PGC1-α下调很可能主要通过转录水平调节的。对于脓毒症性AKI,内毒素升高及炎症因子的释放是其重要特征,其中炎症因子可直接导致PGC1-α转录抑制。Casemayou等[25]在脓毒症细胞模型中发现,炎症因子γ干扰素(IFN-γ)可介导HNF-1β下调从而抑制PGC1-α转录,随后他们在体内实验中也证实肾小管上皮细胞中HNF-1β是PGC1-α的转录因子,过表达/敲除HNF-1β可改善/加重AKI。另外,肿瘤坏死因子样细胞凋亡弱诱导物(tumor necrosis factor-like weak inducer of apoptosis,TWEAK)也可导致PGC1-α转录抑制,AKI时肾小管上皮细胞TWEAK/Fn14表达上调,激活经典NF-κB通路,促进TNF-α释放,从而抑制PGC1-α转录,加剧肾小管细胞凋亡及代偿性增生、重构[26]。然而,尚无研究证实TNF-α具体通过何种转录因子来调节PGC1-α转录。在脓毒症细胞模型中,脂多糖(LPS)是否能够独立介导PGC1-α转录抑制目前尚属未知。但有研究显示,LPS可通过Toll样受体4(TLR4)激活TLR4/丝裂原活化蛋白激酶(MEK)/细胞外调节蛋白激酶(ERK)通路[27]。而在缺血再灌注AKI模型中,活化的ERK可磷酸化FOXO3a导致其入核障碍,抑制PGC1-α转录,从而介导PGC1-α转录下调[28]。基于上述结果推测,LPS可能与肾小管上皮细胞TLR4结合,从而通过MEK/ERK/FOXO3a/PGC1-α通路介导脓毒症性AKI时PGC1-α的转录抑制。
最新研究表明,PGC1-α小分子靶向药物ZLN005在脑缺血神经元损伤、冠心病、糖尿病中具有保护作用[29-31]。肌肉组织中的PGC1-α水平与胰岛素抵抗和2型糖尿病发病情况呈负相关[32],而ZLN005可通过正反馈环路增加PGC1-α在糖尿病db/db小鼠L6细胞中的表达。ZLN005可使线粒体呼吸轻度解偶联,通过提高细胞内ADP/ATP比值,激活AMPK通路,使PGC1-α磷酸化,磷酸化的PGC1-α进而可辅助MEF2增加对PGC1-α自身的转录,从而减轻db/db小鼠胰岛素抵抗并改善糖耐量。然而,PGC1-α小分子靶向药物是否能够预防及治疗AKI尚需相关研究以证实。
小结:PGC1-α是线粒体生物合成及功能的关键调控者,在AKI的发生发展中扮演重要角色,随着其相关机制不断被阐明,以及PGC1-α上游调节物和下游靶标分子不断被发现,PGC1-α有望成为AKI预防及治疗极具价值的靶点。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
辅导员(2020年6期)2020-04-23
派出所工作(2018年4期)2018-09-10
飞碟探索(2016年11期)2016-11-14
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中外医疗(2015年11期)2016-01-04
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
中国当代医药(2015年16期)2015-03-01
西南军医(2015年6期)2015-01-23
