
甲状腺乳头状癌相关基因的生物信息学分析
2021-08-20 05:41谢鋆晖余济春
赣南医学院学报 2021年7期
谢鋆晖,余济春
(1.南昌大学第二附属医院,江西 南昌 330000;2.赣州市肿瘤医院头颈科,江西 赣州 341000)
甲状腺癌(Thyroid carcinoma,THYC)是由甲状腺滤泡细胞发展而来的一种内分泌系统的肿瘤,甲状腺乳头状癌(Papillary thyroid carcinoma,PTC)是最常见的类型(约占80%),它是一种分化型癌[1-2]。近年来,甲状腺癌的死亡率和乳头状甲状腺癌的发病率不断增加,给人们带来很大的困扰。
随着测序技术的发展和RNA-sequencing(RNAseq)测序技术的应用,基于mRNA水平的生物分子在各种疾病的诊断、预后和治疗中得到广泛关注[3]。RNA-seq使我们对基因的转录本及翻译机制有了更加深入的了解,将RNA序列检测基因融合分析应用于已知致病转录本的差异表达等,RNA-seq已成为科研工作中不可或缺的工具[4]。同时,它也是医学生物学(包括肿瘤领域)中重要的手段[5]。SINGH等通过RNA-seq揭示了甲状腺及甲状腺激素的调节机制[6]。QIU等通过转录组测序手段推测PTC进展的部分分子机制,并预测了治疗的生物标志物[7]。
本研究中我们通过GEO数据集的分析初步筛选出癌与正常组织的差异基因,并进一步结合肿瘤基因组图谱(The cancer genome atlas,TGCA)及人蛋白数据库数据发现CDH2、SeprinA1及FN1与甲状腺癌的关系最为密切。现报告如下。
1 材料与方法
1.1 基因表达数据从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)下载GSE64912数据集,共计22例RNA转录组及表达谱分析结果,其中包含18例乳头状甲状腺癌与4例健康对照[8]。
1.2 基因数据前处理通过在线网站iDEP.92(http://bioinformatics.sdstate.edu/idep92/)对下载的基因表达谱进行数据分析。导入表达谱的基因读数(Read Counts)数据进行数据标准化,通过DESeq2软件包实现常规对数log2转换,减少均值相关方差,输出的数据用于后续分析。
1.3 基因差异表达的分析利用R软件的“edgeR”包进行基因差异表达分析,通过其中的函数包构建线性拟合模型,然后比较甲状腺乳头状癌与正常甲状腺组织的基因表达情况。基因差异表达倍数绝对值去对数值(log2|(fold change[FC])|)>2.0且校正后P值(FDR)<0.05的被认定为差异基因。输出的数据导入iDEP中进行数据可视化,绘制柱状图及火山图。
1.4 差异基因的富集分析利用在线网站DAVID(https://david.ncifcrf.gov/)将分析后的差异基因进行基因本体论(Gene Ontology,GO)功能分析,包括生物过程、细胞成分和分子功能,并输出数据。以P<0.05的显著性水平作为截断标准,并使用R软件的“GOplot”包对DAVID富集分析结果进行可视化,绘制和弦图。
1.5 蛋白网络互作及hub基因将筛选出的差异基因输入到基因检索搜寻工具(Search Tool For The Retrieval Of Interacting Genes,STRING)在线数据库(https://string-db.org/)中(置信分数>0.9),分析蛋白间相互作用(Protein-protein Interaction,PPI)。通过Cytoscape软件(3.8.1版本)重建网络图,并利用“cytoHubba”插件识别排名前十(top10)的枢纽基因(Hub genes)。
1.6 统计学方法通过Graph Pad(8.0版本)软件进行student t-test检验来验证核心基因的表达,P<0.05为差异有统计学意义,统计后通过Graph Pad绘制统计图形。
2 结 果
2.1 数据标准化数据库中包含55 637个基因,利用iDEP对数据进行清洗后获得24 566个有效基因,进一步进行数据转化及标准化后进行后续分析(图1A),主成分分析(Principal Component Analysis,PCA)结果提示甲状腺癌与正常甲状腺组织的转录组存在差异(图1B)。

图1 RNA测序数据的标准化.
2.2 基因的差异表达及聚类分析利用limma包对差异基因进行分析,筛选条件:|logFC|>3、adj.p.val<0.05,对筛选出的前160个基因进行层次聚类分析,结果显示可较好的进行聚类(图2A)。甲状腺乳头腺癌(Cancer)与正常甲状腺组织(Normal)相比,其中上调基因100个,下调基因60个(图2B)。

图2 基因的差异表达分析
2.3 差异基因的功能富集分析对上述160个差异基因进行GO分析,条件:P<0.01、基因富集数>3及富集因子>1.5,结果显示前20生物学过程(Biological processes,BP)的主要富集功能,分别为细胞连接组织(Cell junction organization)、突触组织的调节(Regulation of synapse organization)、蛋白激酶活性调节(Regulation of protein kinase activity)、胶原蛋白结合(Synaptic signaling)、兴奋性突触(Excitatory synapse)、细胞基质粘附(Cell-substrate adhesion)、胶原合成(Collagen formation)、gtp酶活性调节(Regulation of gtpase activity)等(图3),基因富集的功能与癌症的发生进展密切相关,进一步提示差异基因在甲状腺癌中的作用。
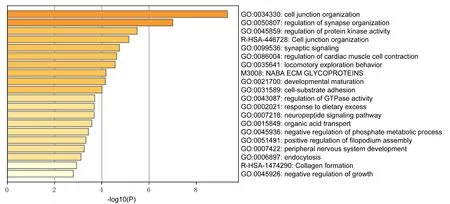
图3 差异基因功能富集结果(Top20)
2.4 分子网络互作筛选关键基因为进一步探究甲状腺癌中关键的潜在靶点,我们将上述差异表达最显著的基因进行蛋白互作分析,获得蛋白-蛋白相互作用(PPI)网络图,包括了97个节点和148个边(图4);进一步确认了处于网络核心区域的基因(Top10)包括:FN1、SerpinA1、PLAU、APOE、TOP2A、CDH2、BUB1、ASPM、MKI67、APOC1,它们所表达的蛋白及信息如表1所示。

图4 蛋白质-蛋白质相互作用(PPI)网络图

表1 Top10关键基因及相关蛋白的信息
2.5 TCGA数据库分析甲状腺癌mRNA表达水平我们通过在线分析网站GEPIA2(gepia.cancerpku.cn)分析TCGA数据库中甲状腺癌的表达数据,通过设定差异表达倍数绝对值对数值(log2|(fold change[FC])|)>2.0,且校正后P值(FDR)<0.05作为统计学差异截断值;进一步证实甲状腺乳头状癌中CDH2、SerpinA1、PLAU、FN1表达较正常甲状腺组织显著上调(P<0.05),ASPM、APOE、APOC1、TOP2A、MKI67、BUB1的表达差异无统计学意义(图5)。
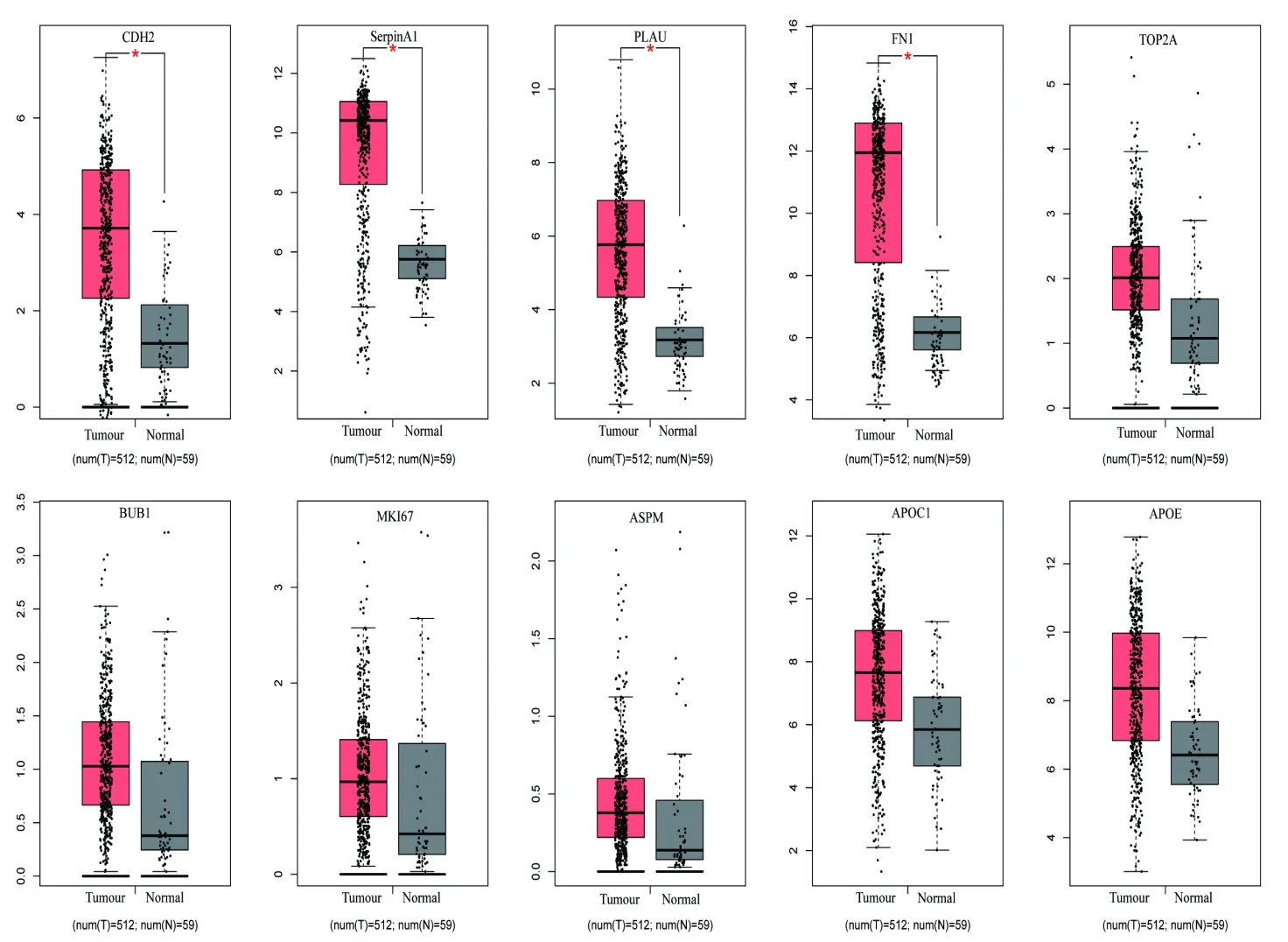
图5 TGCA数据分析基因mRNA的表达情况
2.6 人蛋白质数据库分析关键基因的蛋白质表达我们利用人蛋白质数据库(Human Protein Atlas,HPA)进一步分析甲状腺癌上mRNA具有表达差异的基因(CDH2、SerpinA1、PLAU、FN1)的蛋白表达情况,结果显示,CHD2、SerprinA1及FN1在正常甲状腺组织中弱阳性或阴性表达,在甲状腺乳头状癌中强阳性表达,而PLAU在正常组织及癌组织中均是强阳性表达(图6)。因此,结合mRNA表达数据,最终筛选出CHD2、SerprinA1及FN1为甲状腺癌的关键基因。

图6 HPA数据库分析基因的蛋白表达情况
3 讨论
甲状腺乳头状癌对人们产生了很大的危害,深入研究其分子机制十分必要。本研究旨在利用公共数据库中的mRNA测序数据来确定有助于PTC诊断及研究的分子。我们选取了一项包含18例PTC及4例正常甲状腺组织的研究数据,通过对甲状腺组织的高通量mRNA测序结果进行分析,共筛选了160个差异表达的基因,包括100上调和60下调的基因。对这些差异基因进行功能富集及分析得到如“蛋白质细胞外基质”“钙离子结合”“胶原蛋白结合”等有意义的通路。我们对差异表达最显著的基因进行蛋白互作分析,得到10个关键基因:ASPM、
TOP2A、BUB1、MKI67、APOC1、APOE、CDH2、FN1、SerpinA1、PLAU。通过TGCA数据库分析各个基因的mRNA表达水平及HPA数据库观察蛋白表达水平,我们确定了3个有意义的基因(FN1、SerpinA1、CDH2),可能在甲状腺癌的发生发展过程中扮演重要作用。
纤维连接蛋白1(Fibronectin 1,FN1)一种重要的细胞外基质分子,参与基质重塑、细胞粘附和迁移。NK细胞受体NKp46介导IFN-γ的产生导致肿瘤中FN1的表达增加,改变原发性肿瘤的结构并减少了转移的形成[9]。在食管鳞状细胞癌中FN1上调激活Erk信号进而促进食道癌的进展,高表达FN1与食管癌的生存率低及早期复发相关[10]。FN1在胚胎发生、分化、癌变和转移中也起着至关重要的作用,其可通过PI3K/Akt途径激活MMP9来促进卵巢癌转移[11]。在结肠癌细胞中,纤连蛋白与侵袭能力有关,FN1中的单核苷酸多态性影响结肠癌的肿瘤侵袭、转移[12]。此外,对甲状腺髓样癌的蛋白组学分析也发现FN1可作为新型的预后标记物[13]。但FN1在分化型甲状腺癌中扮演什么角色尚未有明确报道。
SerpinA1基因编码的α-1-抗胰蛋白酶(AAT)是一种主要在人类肝脏中合成的急性期糖蛋白(占80%),是人类的主要血液蛋白[14]。AAT是丝氨酸蛋白酶抑制剂(Serine protease inhibitor,SPI)的一种,通过蛋白酶抑制和非抑制机制调节急性炎症过程,SerpinA1的启动子对IL-6和IL-1途径以及缺氧也有反应。研究表明,AAT的升高可促进癌症的发展[15]。在肺癌、乳腺癌等多种癌症中检测到水平升高,并与预后不良相关[16]。一项肺癌患者血清AAT的分析结果表明,SerpinA1的升高与肿瘤发展有关,并且这种作用不仅仅是因为炎症引起的[17]。目前,SerpinA1在甲状腺癌中的研究仍是空白,SerpinA1基因和AAT蛋白在甲状腺癌的发病机制中是否发挥积极作用,还是仅反映与癌症发展相关的炎症反应,这些问题有待进一步研究和解决。
CDH2基因编码N-钙黏蛋白,具有维持细胞的完整性并参与多种细胞信号转导途径,主要涉及上皮-间充质转换、细胞周期、促血管生成等,参与调控肿瘤的增殖转移,同时也是多种癌症的预后不良标志物[18]。EphB1信号介导TGF-β激活CDH2,并促进肺癌细胞的侵袭和迁移[19]。BACH1通过直接激活CDH2转录来促进上皮-间质转化及血管生成促进食管癌的进展[20]。LncRNA JPX介导miR-944/CDH2信号调控口腔鳞癌细胞的增殖、侵袭及转移等[21]。CDH2除了影响EMT过程外,还与MAPK/Erk、PI3K/Akt和p16/Rb信号通路活化相关,促进甲状腺癌的发生[22]。甲状腺癌具有早期淋巴结转移的特点,CDH2是否在其中发挥作用也需进一步探讨。
通过公共数据的挖掘,基于GEO数据及TGCA数据的生物信息分析结果,筛选出差异表达的基因,预测了部分基因的潜在功能,为后续进一步研究提供依据。因此,我们希望从上述筛选所得的关键基因入手,观察这些关键基因对甲状腺癌细胞的生物学特性,研究其与甲状腺癌相关通路的作用,为临床治疗PTC提供更准确的治疗靶点和奠定新的理论基础,使其有望成为PTC早期诊断及预后的潜在分子标记物。
猜你喜欢
国际放射医学核医学杂志(2021年10期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
心电与循环(2020年1期)2020-02-27
江苏农业科学(2017年5期)2017-04-15
癌症进展(2016年10期)2016-03-20
中国卫生标准管理(2015年2期)2016-01-15
中国病理生理杂志(2015年8期)2015-12-21
中华皮肤科杂志(2014年3期)2014-12-19
湖北农业科学(2014年3期)2014-07-21
中国中西医结合外科杂志(2013年3期)2013-03-11
